
单细胞解析脓毒症免疫通路:fgsea富集分析实战
GSEA富集分析的基本原理是使用预定义的基因集 (例如:铁死亡通路,只要能获取到相关基因,就能定义成一个基因集),将基因按照在两类样本中的差异表达程度进行排序,然后检验这些预先设定的基因集合是否在这个排序表的顶端或者底端富集。如果基因集在排序列表的顶部富集 (即上调),如果在底部富集 (即下调)。
fgsea能够快速对预选基因集进行富集分析,预选基因集可以是自己设定,一般使用MSigdb(Molecular Signatures Database)数据库,同样由提出GSEA方法团队提供。该数据库包含了以下9种不同基因的基因,可供下载以及R软件包载入。
H:hallmark gene sets---癌症特征基因集合,共50 gene sets。
C1:positional gene sets---染色体位置基因集合,共299 gene sets。
C2:curated gene sets---包含了已知数据库,文献等基因集信息,包含5529 gene sets。
C3:motif gene sets---调控靶基因集合,包括miRNA-target以及转录因子-target调控模式,3735 gene sets。
C4:computational gene sets---计算机软件预测出来的基因集合,主要是和癌症相关的基因,858 gene sets。
C5:GO gene sets---Gene Ontology对应的基因集合,10192 gene sets。
C6:oncogenic signatures---致癌基因集合,大部分来源于NCBI GEO发表芯片数据,189 gene sets。
C7:immunologic signatures---免疫相关基因集,4872 gene sets。
C8:single cell identitiy gene sets, 302 gene sets
载入相关R包;
载入数据库基因集;
载入单细胞转录组Seurat对象,获取marker基因,以0群为例,相当于0群与其余各群的差异分析,非单细胞测序数据直接做差异分析。
# 检查msigdbr包是否已安装 if (!requireNamespace("msigdbr", quietly = TRUE)) { install.packages("msigdbr") } # 检查BiocManager包是否已安装 if (!requireNamespace("BiocManager", quietly = TRUE)) { install.packages("BiocManager") } # 使用BiocManager安装fgsea包 if (!requireNamespace("fgsea", quietly = TRUE)) { BiocManager::install("fgsea") } library(msigdbr) #提供MSigdb数据库基因集 library(fgsea) library(dplyr) library(tibble) library(Seurat) library(ggplot2) combined_data_rename1 <- readRDS("D:\\Users\\Desktop\\RDS\\combined_data_rename.rds") # 查看样本分组分布 table(combined_data_rename1@meta.data$sample_group) # 创建新的元数据列,合并细胞类型和样本组信息 combined_data_rename1$celltype.group <- paste(combined_data_rename1$celltype, combined_data_rename1$sample_group, sep = "_") # 确保细胞类型标识正确 combined_data_rename1$celltype <- Idents(combined_data_rename1) # 查看新创建的分组分布 table(combined_data_rename1$celltype.group)

# 设置Seurat对象的活动标识为新的分组 Idents(combined_data_rename1) <- "celltype.group" # 执行差异表达分析 # FindMarkers参数说明: # ident.1 = 'NK_S':比较的第一组(处理组,此处为脓毒症NK细胞) # ident.2 = 'NK_NS':比较的第二组(对照组,此处为非脓毒症NK细胞) # min.pct = 0.1:基因至少在10%的细胞中表达 # logfc.threshold = 0:不设置logFC阈值,返回所有差异 markers <- FindMarkers( combined_data_rename1, ident.1 = 'NK_S', ident.2 = 'NK_NS', min.pct = 0.1, logfc.threshold = 0 )
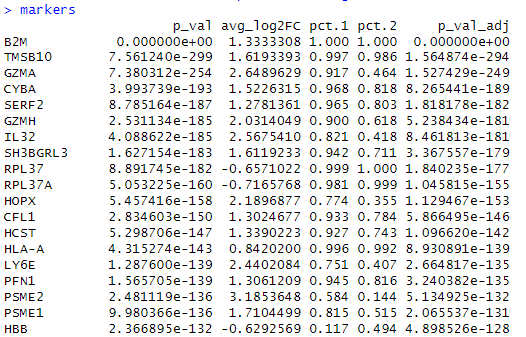
# 为markers数据框添加基因名列 markers$genes = rownames(markers) # 准备GSEA排名列表 # 按平均log2FC降序排列,选取基因名和log2FC两列 cluster.genes <- markers %>% arrange(desc(avg_log2FC)) %>% dplyr::select(genes, avg_log2FC) # 将数据框转换为命名向量(fgsea所需格式) # 基因名作为名称,log2FC作为值 ranks <- deframe(cluster.genes) # 加载KEGG通路基因集 # msigdbr参数说明: # species = "Homo sapiens":使用人类基因集 # category = "C2":选择C2类别(包括KEGG、BIOCARTA、REACTOME等) mdb_c2 <- msigdbr(species = "Homo sapiens", category = "C2") # 筛选KEGG通路并转换为fgsea所需格式 # 创建列表,通路名为名称,包含的基因向量为值 fgsea_sets = mdb_c2[grep("^KEGG", mdb_c2$gs_name),] %>% split(x = .$gene_symbol, f = .$gs_name) # 查看加载的通路数量 length(fgsea_sets) # 执行fgsea分析 # fgsea参数说明: # pathways = fgsea_sets:基因集列表 # stats = ranks:基因排名向量 # nperm = 1000:排列检验次数(通常设置为1000-10000) fgseaRes <- fgsea(fgsea_sets, stats = ranks, nperm = 1000) # 查看fgsea结果 fgseaRes
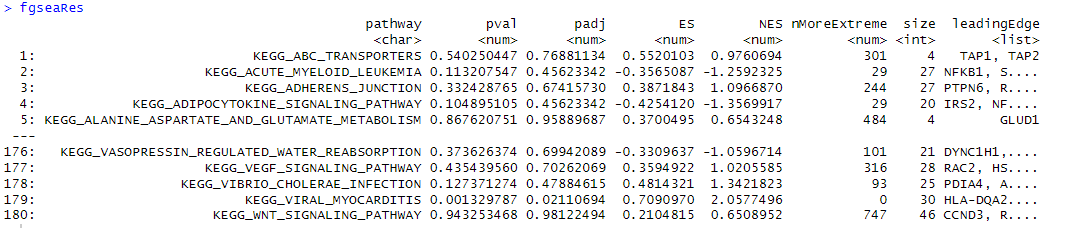
fgsea结果解读:
pathway: 通路的名称,例如KEGG_ABC_TRANSPORTERS。
pval: 富集分析的p值,表示该通路富集的显著性。p值越小,表示该通路富集越显著。注意,这里使用的是基于置换检验(permutation)得到的p值。
padj: 调整后的p值,通常使用Benjamini-Hochberg方法对p值进行多重检验校正。padj小于0.05通常被认为是显著的。
ES: 富集分数(Enrichment Score)。它是FGSEA分析的核心指标,表示基因集中基因在排序列表中的富集程度。正值表示基因集在排序列表的顶部富集(即上调),负值表示在底部富集(即下调)。
NES: 标准化后的富集分数(Normalized Enrichment Score)。它对ES进行了标准化,使得不同基因集之间的富集分数可以进行比较。通常,NES的绝对值越大,表示富集程度越高。
nMoreExtreme: 在置换检验中,得到比当前观察到的ES更极端的ES值的次数。这个值用于计算p值(即nMoreExtreme / nPerm)。
size: 该通路基因集中基因的数量。
leadingEdge: 核心基因集,即对富集分数贡献最大的基因列表。这些基因通常位于排序列表的顶部或底部,是驱动通路富集的关键基因。
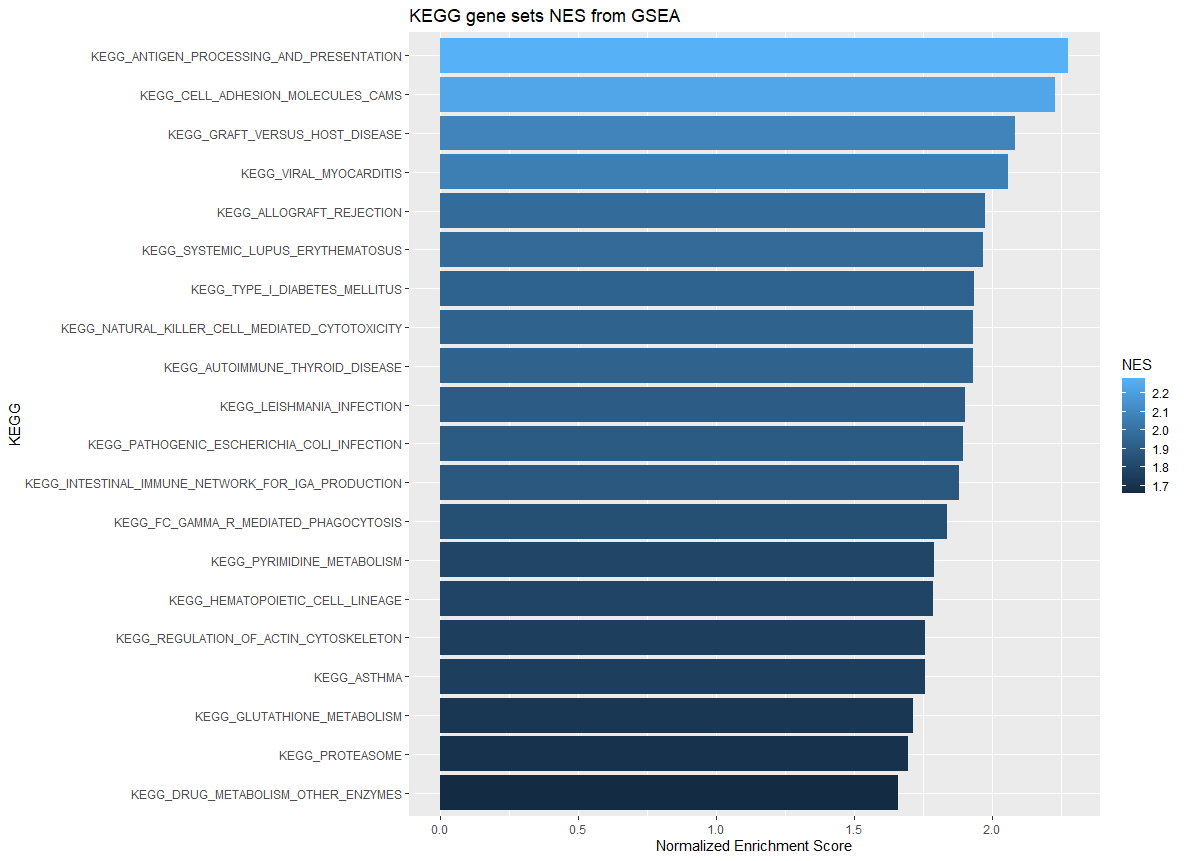
# 先转换为数据框,处理leadingEdge列为字符串 fgseaRes_df <- as.data.frame(fgseaRes) fgseaRes_df$leadingEdge <- sapply(fgseaRes_df$leadingEdge, function(x) paste(x, collapse = ",")) write.csv(fgseaRes_df, file = "D:\\Users\\Desktop\\fgsea_results.csv", row.names = FALSE) # 绘制GSEA结果条形图 # 筛选p值<0.05的显著通路,按NES降序排列,取前20个 p <- ggplot( fgseaRes %>% as_tibble() %>% arrange(desc(NES)) %>% filter(pval < 0.05) %>% head(n = 20), aes(reorder(pathway, NES), NES) ) + geom_col(aes(fill = NES)) + coord_flip() + labs( x = "KEGG Pathway", y = "Normalized Enrichment Score", title = "KEGG Pathway Enrichment Analysis (Top 20)" ) + theme_minimal() # 保存条形图 pdf('D:\\Users\\Desktop\\脓毒症单细胞\\RESULT\\GSEA-fgsea.pdf', width = 8, height = 5) print(p) dev.off()
如图所示,NES为每个基因子集ES根据基因集大小标准化的富集分数,ES反应基因集在排序列表两端的富集程度。
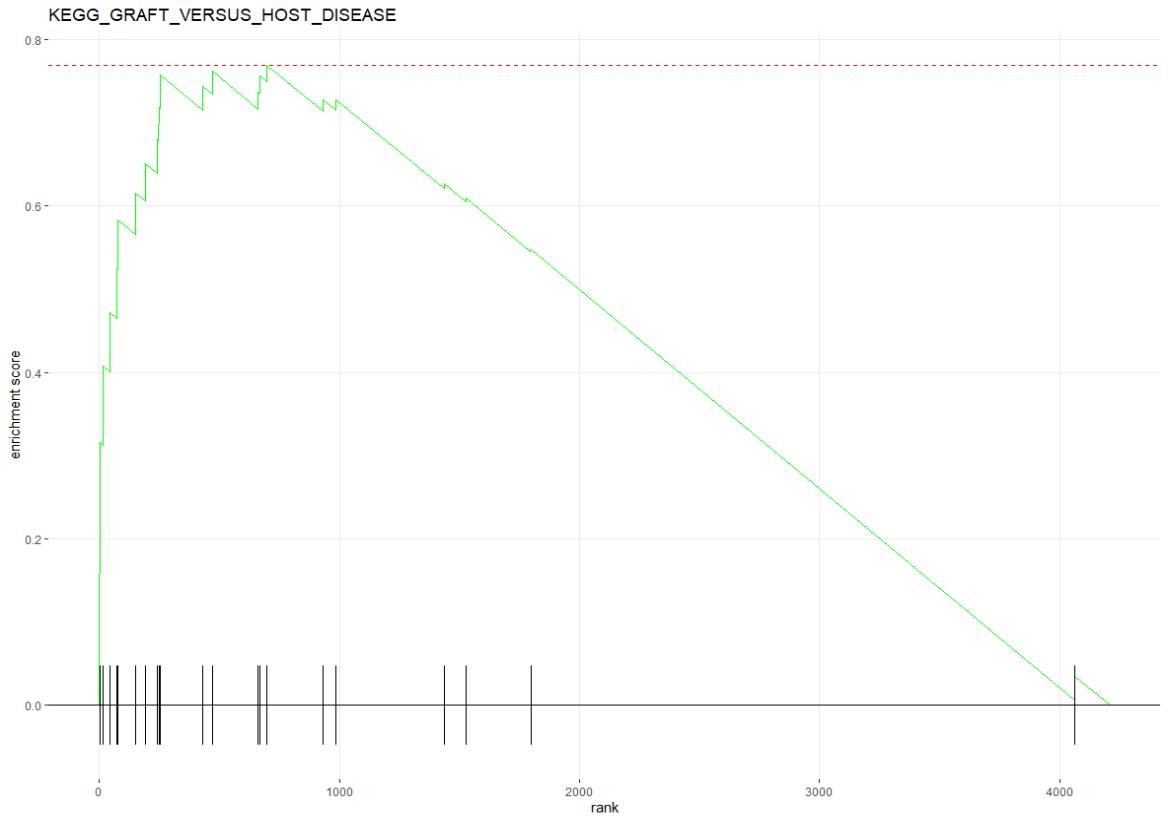
# 绘制特定通路富集图 # 绘制"KEGG_GRAFT_VERSUS_HOST_DISEASE"通路富集曲线 pdf('D:\\Users\\Desktop\\脓毒症单细胞\\RESULT\\fgsea_KEGG_PRIMARY_IMMUNODEFICIENCY.pdf', width = 8, height = 5) plotEnrichment(fgsea_sets[["KEGG_GRAFT_VERSUS_HOST_DISEASE"]], ranks) + labs(title = "KEGG_GRAFT_VERSUS_HOST_DISEASE") dev.off()
如下图,展示了其中KEGG_GRAFT_VERSUS_HOST_DISEASE通路结果,横坐标从大到小排序好的所有基因,靠近0的基因代表表达量与表型呈现正相关,远离0的为负相关基因。纵坐标纵坐标是富集打分ES,这个富集打分是一个动态的值,通常用偏离0最远的值作为富集打分。 中间的竖线为感兴趣基因集所处的位置。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号