RNA-seq数据分析实战:基于DESeq2的差异表达基因鉴定与结果解读
在转录组分析中,差异分析是必不可少的一步。那什么是差异分析呢?差异分析的结果又该怎么解读?今天就让我们一起来深入了解一下,同时认识与它紧密相关的几个关键指标,以及怎么做差异分析。
1. 转录组差异分析基础知识
1.1 什么是转录组?
简单来说,转录组是指一个活细胞在特定时间和环境下,所能转录出来的所有RNA的总和。因此,转录组分析就是为了了解基因的表达调控机制和功能。
1.2 差异分析是什么?为什么要做差异分析?
转录组差异分析是一种生物信息学分析方法,旨在比较不同样本或条件下转录组数据的差异,以揭示基因表达的变化。从所有基因中找出不同样本中表达具有差异的基因,了解这些差异基因如何影响生物体的生理功能和疾病发生发展。
2. 差异分析(基于DESeq2)
2.1. 数据准备
我们使用示例的表达矩阵进行差异分析演示。
# 加载必要的包
library(DESeq2)
library(dplyr)
library(tibble)
导入原始基因表达量count表达矩阵:
dat <- read.csv("count.csv",check.names = F,row.names = 1,header = T)
基因表达值矩阵:行是基因,列是样本,内容为基因表达值,表达矩阵的样本列;
转录组分析原始基因表达量计数数据:
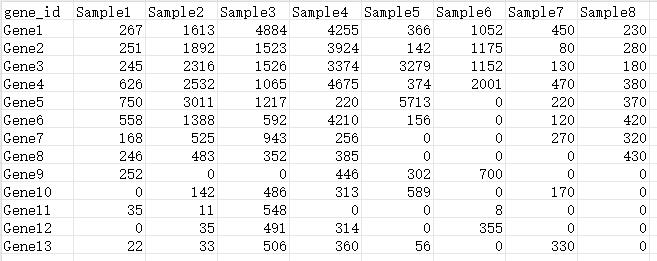
注意第一列基因名称不能有重复值。DESeq2只接受未经标准化处理的转录组分析原始基因表达量计数数据。
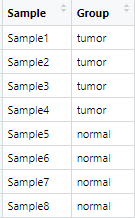
导入分组信息:
sample_groups <- read.csv("group.csv",check.names = F,row.names = 1,header = T) # 设置参考水平(对照组normal在前) sample_groups$Group <- factor(sample_groups$Group, levels = c("normal", "tumor"))
分组信息表如下图所示,第一列为样本ID,第二列为样本分组:
# 创建DESeqDataSet dds <- DESeqDataSetFromMatrix( countData = dat, colData = sample_groups, design = ~ Group ) # 过滤低表达基因(保留至少在3个样本中count >= 10的基因) keep <- rowSums(counts(dds) >= 10) >= 3 dds <- dds[keep, ] # 运行DESeq2 dds <- DESeq(dds) # 获取差异分析结果 results <- results(dds, contrast = c("Group", "tumor", "normal")) # 将结果转换为数据框并添加基因名 res_df <- as.data.frame(results) %>% tibble::rownames_to_column("Gene") %>% dplyr::arrange(padj) # 按校正p值排序 # 定义差异基因筛选阈值 lfc_cutoff <- 1 # log2倍变化阈值 padj_cutoff <- 0.05 # 校正p值阈值 # 筛选显著差异基因 sig_genes <- res_df %>% dplyr::filter( !is.na(padj), abs(log2FoldChange) >= lfc_cutoff, padj <= padj_cutoff ) # 添加差异表达方向列 sig_genes <- sig_genes %>% dplyr::mutate( Regulation = dplyr::case_when( log2FoldChange >= lfc_cutoff & padj <= padj_cutoff ~ "Up", log2FoldChange <= -lfc_cutoff & padj <= padj_cutoff ~ "Down", TRUE ~ "Not Significant" ) )
baseMean: 基因在所有样本中的平均表达量,反映基因的整体表达水平
log2FoldChange: 基因表达的对数2倍变化值(以2为底),正值表示在疾病组中表达上调,负值表示下调
lfcSE: log2FoldChange的标准误,反映fold change估计的精确度
stat: Wald检验统计量,用于评估表达变化的显著性程度
pvalue: 原始p值,表征差异表达的统计显著性(未校正多重假设检验)
padj: 经Benjamini-Hochberg法校正后的p值,用于控制错误发现率(FDR)
Regulation: 基因表达调控方向,基于log2FoldChange和padj的筛选结果判定
"Up": 在疾病组中显著上调(log2FC ≥ 1且padj ≤ 0.05),"Down": 在疾病组中显著下调(log2FC ≤ -1且padj ≤ 0.05),"Not Significant": 差异表达未达到显著性阈值
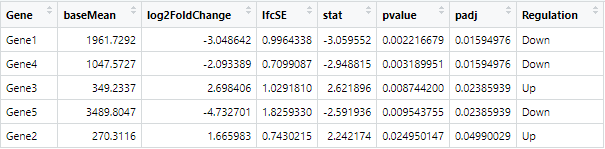
保存完整结果
write.csv(res_df, "DESeq2_All_Genes_Results.csv", row.names = FALSE)
# 保存显著差异基因结果
write.csv(sig_genes, "DESeq2_Significant_DEGs.csv", row.names = FALSE)

 浙公网安备 33010602011771号
浙公网安备 33010602011771号