转录组基因表达差异分析全流程:以GSE65682为例
在转录组分析中,差异分析是必不可少的一步。那什么是差异分析呢?差异分析的结果又该怎么解读?以《GEO数据库转录组芯片数据处理与R分析:以GSE65682为例》一文中的数据集(GSE65682)为例,今天就让我们一起来深入了解一下,同时认识与它紧密相关的几个关键指标与图形,以及怎么做差异分析。
1. 转录组差异分析基础知识
1.1 什么是转录组?
简单来说,转录组是指一个活细胞在特定时间和环境下,所能转录出来的所有RNA的总和。因此,转录组分析就是为了了解基因的表达调控机制和功能。
1.2 差异分析是什么?为什么要做差异分析?
转录组差异分析是一种生物信息学分析方法,旨在比较不同样本或条件下转录组数据的差异,以揭示基因表达的变化。从所有基因中找出不同样本中表达具有差异的基因,了解这些差异基因如何影响生物体的生理功能和疾病发生发展。
1.3 差异分析结果解读

行名为基因名称,logFC表示基因在两个不同条件(例如处理组与对照组)之间的表达倍数的对数值。正值表示该基因为上调基因(处理组相对于对照组),负值表示该基因为下调基因。AveExpr表示该基因在所有样本中表达水平的平均值。PValue是统计学检验变量,代表差异显著性,一般pvalue小于0.05代表具有显著性差异,但可根据具体情况适当调整。adj.P.Val为校正后的P值,通过FDR校正调整原始P值,以控制多重比较中的假阳性率。校正后的P值较小的基因更有可能是真正的差异表达基因。singnificant代表差异显著性标志,down表示下调基因,up表示上调基因,no表示无显著性差异基因。
1.4 如何判断一个基因是否是差异基因呢?
通常会综合考虑logFC、pvalue或adj.P.Val这三个指标。一般来说,一个基因要被判定为差异基因,需要同时满足logFC符合设定的阈值(如logFC绝对值大于2)、pvalue小于0.05或adj.P.Val小于0.05。当然这些阈值要根据实际情况做相应的调整,常见的logFC会选择1,1.2,1.5等,pvalue常见的有0.05,0.01等。
注:pvalue和adj.P.Val选择一个就行了,从严格意义上讲选择adj.P.Val会更好,但是有时候选adj.P.Val的话,一个差异基因也筛选不出来,要根据实际情况做相应的变动
1.5 用火山图呈现差异结果
做完差异分析后通常要做火山图,来呈现差异分析的结果。
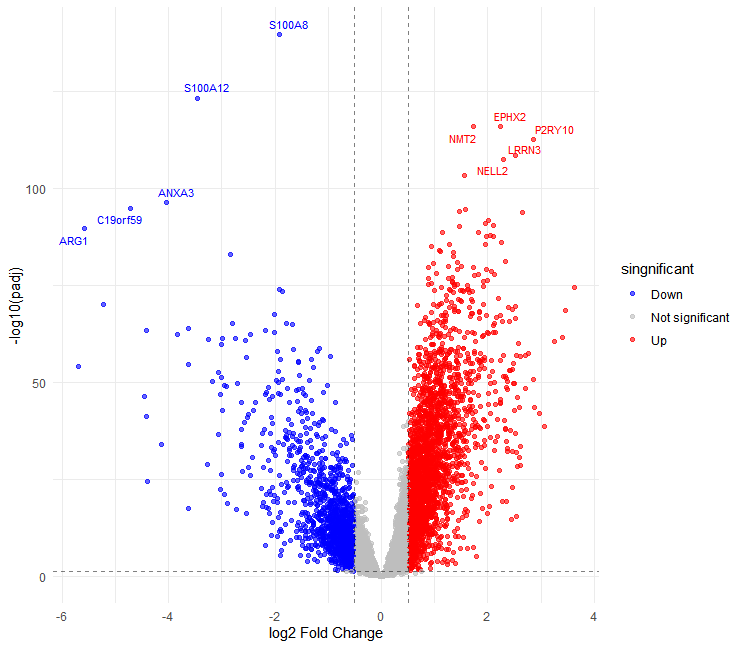
火山图是一种常用的可视化工具,它以logFC值为横坐标,以 -log10(padj)为纵坐标。在火山图中,每个点代表一个基因,横坐标表示基因表达的变化倍数,纵坐标表示差异的显著性。左上角蓝色区域为下调基因,右上角红色区域为上调基因,中间灰色区域为无显著性差异基因。通过火山图,我们可以快速直观地识别出哪些基因是显著差异基因,以及它们是上调还是下调。
2. 差异分析(基于limma)
2.1. 数据准备
我们将使用《GEO数据库转录组芯片数据处理与R分析:以GSE65682为例》一文中数据处理后的表达矩阵进行差异分析演示。
# 加载必要的包 library(limma) library(dplyr) 导入处理好的表达矩阵: expr_data <- read.csv("GSE65682_exp_unique.csv", row.names = 1)
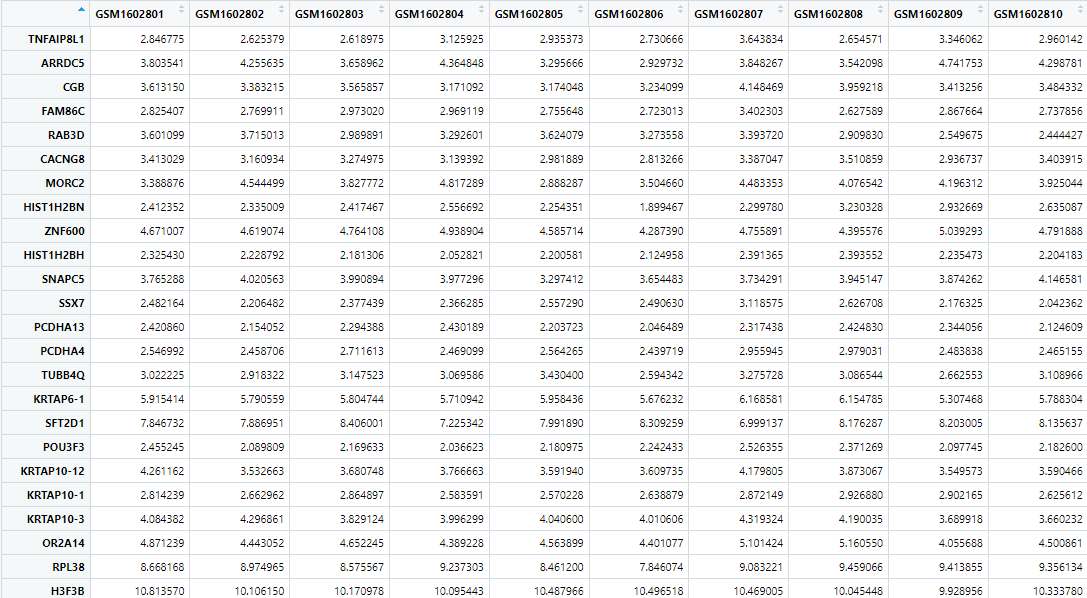
表达矩阵如下图所示,每一行是一个基因,每一列是一个样本,中间数字的是基因表达量。
导入处理好的分组信息表:
group <- read.csv("GSE65682_group.csv") sample_group <- group %>% mutate(group = ifelse(group == "Alive", "Sepsis", ifelse(group == "Dead", "Sepsis","Healthy"))) group_info <- sample_group
样本分组数据包含样本名称和分组信息。第一列样品名称(sample),第二列为分组名称(group)。

提取表达矩阵和分组因子
# 从 group_info 中提取唯一的样本名称/标识符 sample_ids <- unique(group_info$sample) # 提取 expr_data 中对应的列 expr_data_subset <- expr_data[, colnames(expr_data) %in% sample_ids] # 从 group_info 中提取分组因子 group_factor <- factor(group_info$group[match(colnames(expr_data_subset), group_info$sample)])
根据匹配的表达矩阵和分组因子,进行 limma 分析
# 创建设计矩阵 design <- model.matrix(~0 + group_factor) colnames(design) <- levels(group_factor) # 将表达矩阵转换为 voom 对象 # v <- voom(expr_data_subset, design) # 拟合线性模型,这次使用正确的数值型矩阵 fit <- lmFit(expr_data_subset, design) # 设置对比(假设有两个组:group1 和 group2) cont.matrix <- makeContrasts(Healthy - Sepsis, levels = design) # 进行对比测试 fit2 <- contrasts.fit(fit, cont.matrix) fit2 <- eBayes(fit2) # 获取结果 results <- topTable(fit2,n = Inf, adjust = "fdr") ## 导出所有的差异结果 nrDEG = na.omit(results) ## 去掉数据中有NA的行或列 diffsig <- nrDEG # write.csv(diffsig, "all.limmaOut.csv") ## 我们使用|logFC| > 0.5,padj < 0.05(矫正后P值) foldChange = 0.5 padj = 0.05 ## 筛选出所有差异基因的结果 All_diffSig <- diffsig[(diffsig$adj.P.Val < padj & (diffsig$logFC>foldChange | diffsig$logFC < (-foldChange))),] write.csv(All_diffSig, "GSE65682_all.filterdegs.csv") ##输出差异基因数据集
在得到差异分析的结果后,我们接下来使用火山图进行可视化
# 加载必要的可视化包 library(ggplot2) library(ggrepel) library(ggthemes) # 准备火山图数据 volcano_data <- nrDEG volcano_data$gene <- rownames(volcano_data) # 添加差异表达状态列 volcano_data$singnificant <- "NO" volcano_data$singnificant[volcano_data$logFC > foldChange & volcano_data$adj.P.Val < padj] <- "UP" volcano_data$singnificant[volcano_data$logFC < -foldChange & volcano_data$adj.P.Val < padj] <- "DOWN" # 选择要标记的基因(前10个上调和下调基因) top_up <- volcano_data %>% filter(singnificant == "UP") %>% arrange(adj.P.Val) %>% head(5) top_down <- volcano_data %>% filter(singnificant == "DOWN") %>% arrange(adj.P.Val) %>% head(5) top_genes <- rbind(top_up, top_down) volcano_plot <- ggplot(volcano_data, aes(x = logFC, y = -log10(adj.P.Val), color = singnificant)) + geom_point(alpha = 0.6, size = 1.5) + scale_color_manual(values = c("DOWN" = "blue", "NO" = "grey", "UP" = "red"), labels = c("Down", "Not significant", "Up")) + geom_vline(xintercept = c(-foldChange, foldChange), linetype = "dashed", alpha = 0.5) + geom_hline(yintercept = -log10(padj), linetype = "dashed", alpha = 0.5) + geom_text_repel(data = top_genes, aes(label = gene), size = 3, max.overlaps = 20, show.legend = FALSE) + labs(x = "log2 Fold Change", y = "-log10(padj)") + theme_minimal() + theme(legend.position = "right") print(volcano_plot) # 保存火山图 ggsave("volcano_plot.png", volcano_plot, width = 10, height = 8)
火山图结果:


 浙公网安备 33010602011771号
浙公网安备 33010602011771号