

Nature Genetics | 本周最新文献速递
文章标题: Computationally efficient meta-analysis of gene-based tests using summary statistics in large-scale genetic studies
中文标题: 基因研究新利器:REMETA实现大规模基因组关联研究高效整合!
关键词: 基因组关联分析、Meta分析、基因组学、统计方法、计算效率
摘要总结:
基于单变异汇总统计量的基因组测试Meta分析是遗传关联研究的有力策略。然而,现有方法需要为每个研究和感兴趣的性状共享变异之间的协方差矩阵,这在大规模研究中计算、存储和共享都非常繁琐。这篇文章提出了REMETA——一种高效的基因组测试Meta分析工具,它通过为每个研究使用单个稀疏协方差参考文件,并利用单变异汇总统计量对每个性状进行重新缩放,从而解决了这一挑战。
REMETA开发了新的二元性状Meta分析方法,以解决病例-对照不平衡问题,并估算负担测试的等位基因频率、基因型计数和效应大小。研究通过对英国生物样本库(UK Biobank)中五种性状的Meta分析,验证了REMETA的性能和优势。结果显示,REMETA在控制I型错误率方面表现出卓越的准确性,在多种模拟情景下,其统计功效与基于个体水平数据的联合分析相当,同时显著降低了计算成本和存储需求。REMETA还开发了一种紧凑的每染色体二进制文件格式,用于高效存储和共享每项研究所需的LD矩阵,进一步提高了计算效率。这项工作不仅提供了一个可扩展且准确的基因组学Meta分析框架,能够促进大规模外显子组测序研究的整合,对于推动复杂疾病的遗传学研究和新药靶点的发现具有重要意义。
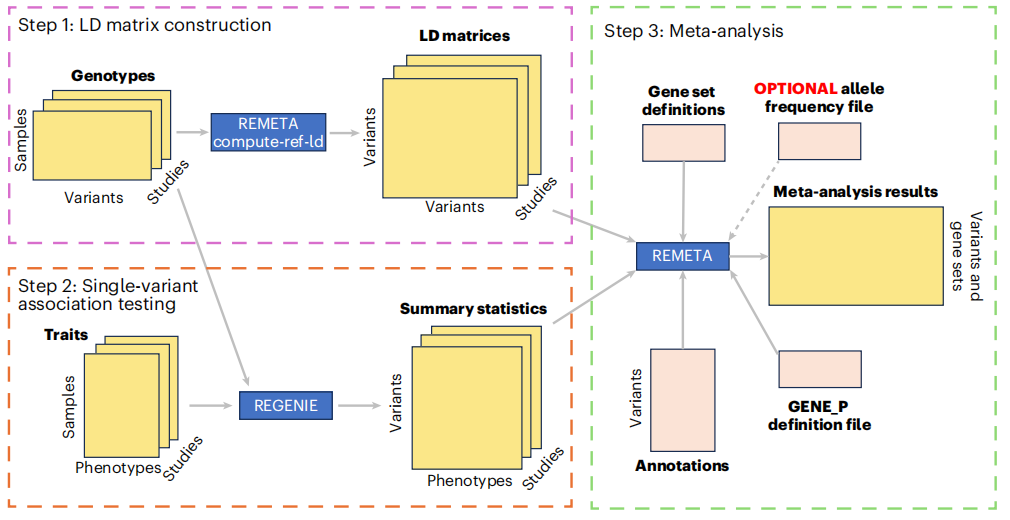
文章的亮点:
- 提出了REMETA,一种高效的基因组测试Meta分析工具,解决了现有方法在处理大规模遗传研究中协方差矩阵计算和存储的挑战。
- 开发了新的二元性状Meta分析方法,有效控制病例-对照不平衡下的I型错误率。
- 实现了计算效率的显著提升,通过使用单个稀疏协方差参考文件和单变异汇总统计量重新缩放LD矩阵,减少了计算成本和存储需求。
- 在英国生物样本库中对五种性状的Meta分析中,REMETA表现出与个体水平数据联合分析相当的统计功效。
- 提供了估算负担测试的等位基因频率、基因型计数和效应大小的方法,增强了结果的可解释性。
文章的局限:
- REMETA方法需要REGENIE软件计算单变异汇总统计量,且任何对表型或协变量的更新都需要重新计算这些统计量。
- 在多祖先群体Meta分析中,如果LD矩阵在不同祖先群体间存在异质性,可能需要更精细的祖先群体特异性LD信息。
- 研究中对某些性状(如LDL)的条件分析结果与REGENIE存在差异,可能需要进一步探究原因。
文章标题: Scalable and accurate rare variant meta-analysis with Meta-SAIGE
中文标题: 稀有变异Meta分析新突破:Meta-SAIGE实现大规模数据高效整合!
关键词: 稀有变异、Meta分析、基因组关联研究、统计方法、计算效率
摘要总结:
Meta分析通过整合多个队列的汇总统计量,增强了稀有变异关联测试的功效。然而,现有方法在处理低流行率二元性状时,往往无法有效控制I型错误率,并且计算成本高昂。这篇文章引入了Meta-SAIGE——一种可扩展且准确的稀有变异Meta分析方法,它能够精确估计零分布以控制I型错误率,并跨表型重用连锁不平衡(LD)矩阵以提高表型组规模分析的计算效率,这对于识别与人类疾病和性状相关的稀有遗传变异具有重要意义。
研究通过英国生物样本库(UK Biobank)和All of Us全外显子组测序数据的模拟研究表明,Meta-SAIGE能够有效控制I型错误率,并达到与SAIGE-GENE+个体水平分析相当的功效。在对UK Biobank和All of Us数据中83种低流行率表型进行Meta分析时,Meta-SAIGE识别出237个基因-性状关联,其中80个在单个数据集中并未达到显著性,突显了Meta分析的强大功效。Meta-SAIGE通过两级鞍点近似(SPA)方法,针对病例-对照不平衡问题进行了I型错误率的调整,显著优于其他方法。此外,Meta-SAIGE在计算成本和存储需求方面也表现出显著优势,能够大幅减少大规模表型组分析的计算负担。这项研究不仅提供了一个可扩展且准确的稀有变异Meta分析框架,还为大规模基因组学研究提供了新的工具,对于推动复杂疾病的遗传学研究和新药靶点的发现具有重要意义。
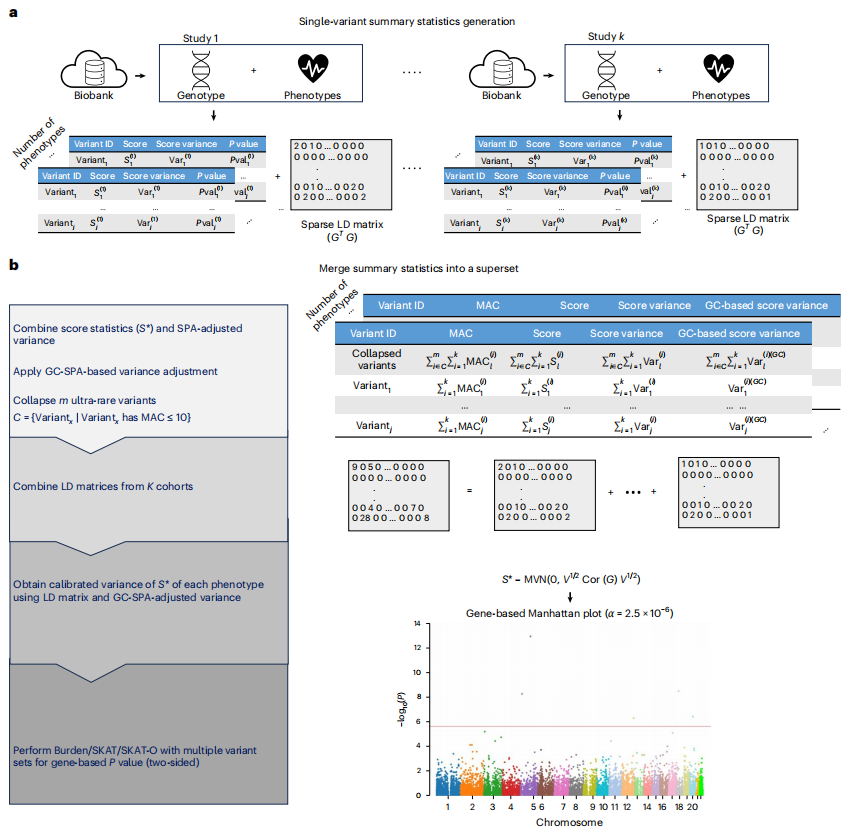
文章的亮点:
- 引入了Meta-SAIGE,一种可扩展且准确的稀有变异Meta分析方法,有效控制I型错误率。
- 通过跨表型重用LD矩阵,显著提高了表型组规模分析的计算效率。
- 在英国生物样本库和All of Us数据中识别出237个基因-性状关联,其中大部分在单个数据集中不显著,凸显了Meta分析的功效。
- 在控制I型错误率和统计功效方面,Meta-SAIGE表现出与个体水平分析相当的性能。
- 提供了估算负担测试的等位基因频率、基因型计数和效应大小的方法,增强了结果的可解释性。
文章的局限:
- Meta-SAIGE在处理特异性性状定义的子集时,如果LD矩阵与全队列数据存在差异,可能会导致结果不一致。
- 研究主要关注欧洲祖先群体,对于其他祖先群体的多祖先Meta分析仍需进一步探索。
- 在某些情况下,Meta-SAIGE的I型错误率控制可能略显保守。
- 对于某些罕见变异,尤其是在小样本量队列中,Meta-SAIGE的功效可能仍受限。
- 研究中使用的模拟数据可能无法完全捕捉真实世界遗传数据的复杂性。
文章标题: Liability threshold model-based disease risk prediction based on electronic health record phenotypes
中文标题: 电子健康档案新洞察:LTPI模型精准预测疾病风险!
关键词: 疾病风险预测、电子健康档案、责任阈值模型、遗传关联、表型整合
摘要总结:
电子健康档案(EHR)作为基因组研究的宝贵资源日益普及,但其临床数据的病例-对照标记往往不准确,导致下游分析效果不佳。这篇文章提出了一种基于责任阈值模型(LTPI)的疾病风险预测方法,它通过结合遗传相关性与EHR表型数据(包括二元和连续性状、诊断代码、家族疾病史、实验室测量和生物标志物),推导出新的连续性状表型,这对于提高疾病风险预测的准确性和全基因组关联研究(GWAS)的功效具有重要意义。
LTPI模型采用自动性状选择算法(ATSA),能够识别最有助于提高风险预测准确性的非目标性状子集,并提供与目标疾病相关的非目标性状见解。通过对eMERGE网络和英国生物样本库(UK Biobank)数据的模拟和应用,研究证明LTPI在疾病风险预测和GWAS功效方面,相比传统表型代码、仅包含家族史的模型以及SoftImpute表型插补方法,均表现出持续的性能提升,同时有效控制假阳性率。LTPI模型能够将二元表型转化为连续风险评分,即使在数据缺失的情况下也能工作,并且能够区分遗传和环境性状相关性,这在传统机器学习方法中是难以实现的。这项研究不仅为利用EHR数据进行疾病风险预测和GWAS分析提供了强大且灵活的工具,对于实现精准医疗和个性化治疗策略具有重要意义。
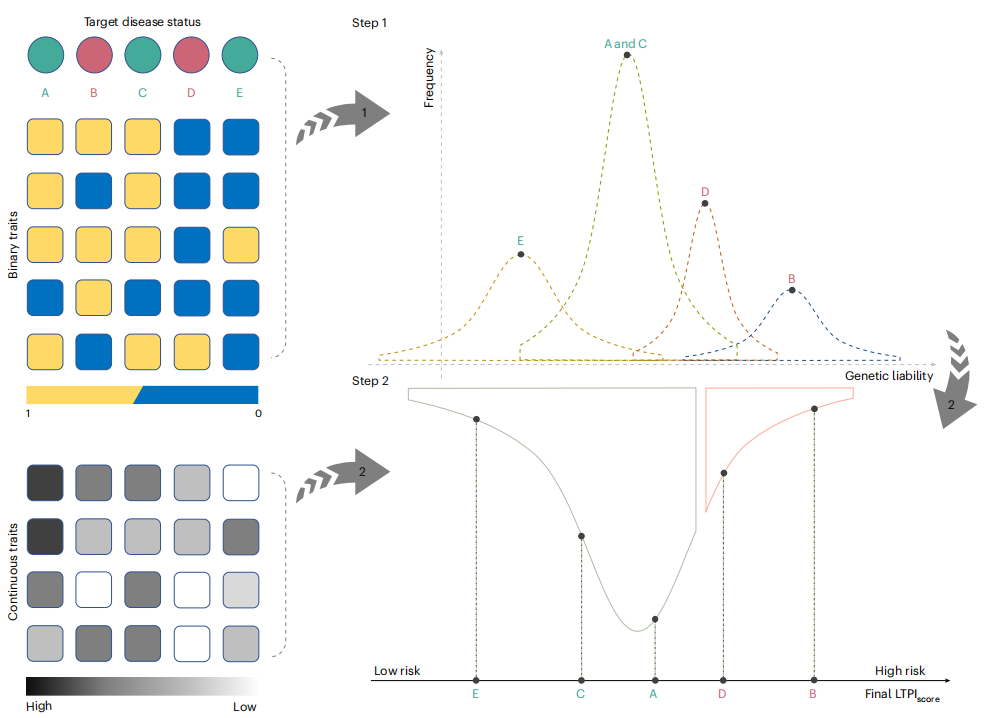
文章的亮点:
- 提出了LTPI模型,一种基于责任阈值模型的新型疾病风险预测方法,有效整合EHR中的丰富表型信息。
- 引入了自动性状选择算法(ATSA),能够智能选择最具信息量的非目标性状,提高预测准确性。
- 在eMERGE网络和英国生物样本库数据中,LTPI在疾病风险预测和GWAS功效方面均优于传统方法。
- LTPI模型能够将二元表型转化为连续风险评分,即使在数据缺失的情况下也能工作,并区分遗传和环境性状相关性。
- 为临床风险分层和基因组关联研究提供了新的工具,有助于精准医疗的发展。
文章的局限:
- 表型预测评估缺乏金标准标签,依赖于不完善的PheCode标签,可能影响预测准确性的评估。
- LTPI模型在处理EHR数据中“非随机缺失”(MNAR)情况时,仍需进一步研究和改进。
- 研究主要关注欧洲人群,其结果在其他祖先群体中的普适性仍需验证。
- LTPI模型在处理遗传相关性较低的非目标性状时,功效提升可能有限。
- 尽管LTPI在计算上是高效的,但其计算复杂性仍可能随性状数量的增加而增加。
文章标题: Genome-wide association study and polygenic risk prediction of hypothyroidism
中文标题: 甲状腺功能减退症:全基因组关联研究揭示疾病风险新维度!
关键词: 甲状腺功能减退症、全基因组关联研究、多基因风险评分、免疫相关基因、疾病进展预测
摘要总结:
原发性甲状腺功能减退症是一种常见且隐匿的代谢性疾病,其遗传基础复杂且异质性高。这篇文章通过对113,393例甲状腺功能减退症病例和1,065,268例对照的欧洲人群进行全基因组Meta分析,并结合甲状腺激素水平数据,识别出350个与甲状腺功能减退症相关的新基因座,其中179个是首次报道,这对于深入理解甲状腺功能减退症的病理生理学和开发新的诊断及治疗策略具有重要意义。
研究发现,许多甲状腺功能减退症风险基因座调控血细胞计数和循环炎症小体,通过多种基因定位策略,优先识别出259个富含免疫相关功能的推定致病基因。研究开发了一种基于超过115,000例甲状腺功能减退症病例的多基因风险评分(PRS),旨在解决甲状腺激素缺乏个体诊断的挑战。结果显示,将PRS与甲状腺激素和甲状腺过氧化物酶自身抗体结合,可实现甲状腺功能减退症最高的预测准确性,并且该PRS能够对亚临床甲状腺功能减退症患者的疾病进展风险进行分层。高风险PRS组的患者在10年内进展为显性甲状腺功能减退症的风险显著升高。这项研究不仅揭示了甲状腺功能减退症的复杂多基因性质和免疫相关机制,还为甲状腺功能减退症的精准诊断、风险分层和潜在治疗靶点提供了新的工具,对于改善患者的预后和开发个性化治疗方案具有重要意义。
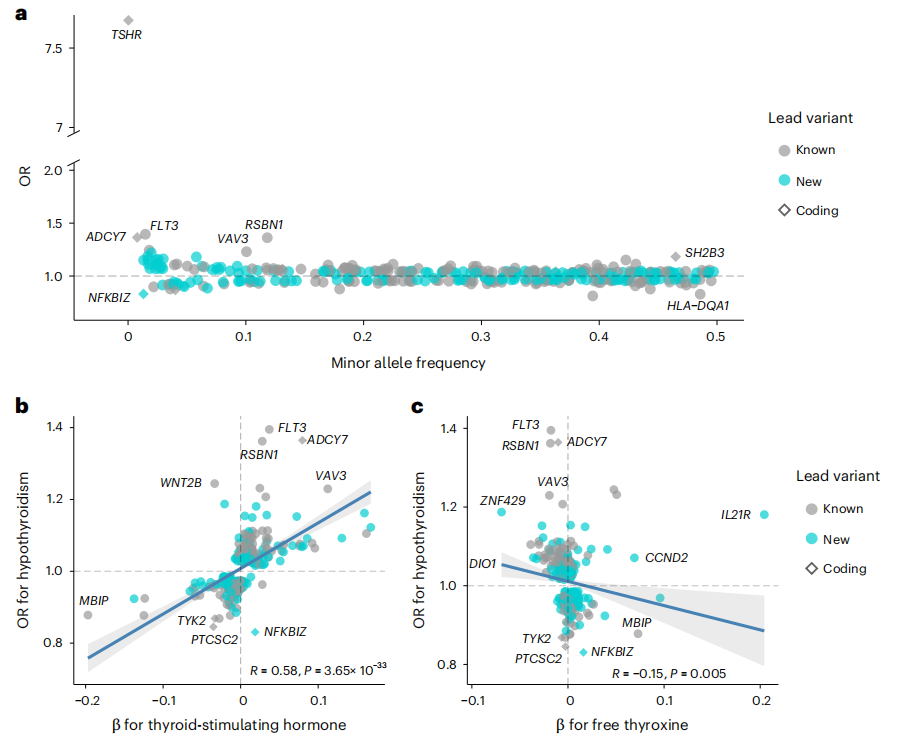
文章的亮点:
- 识别出350个与甲状腺功能减退症相关的新基因座,其中179个是首次报道,显著扩展了对该疾病遗传基础的理解。
- 发现许多甲状腺功能减退症风险基因座调控血细胞计数和循环炎症小体,并优先识别出富含免疫相关功能的致病基因。
- 开发了基于超过115,000例病例的多基因风险评分(PRS),提高了甲状腺功能减退症的预测准确性。
- 证明PRS能够有效对亚临床甲状腺功能减退症患者的疾病进展风险进行分层。
- 揭示了TYK2和ZAP70等基因的常见和罕见蛋白改变变异与甲状腺功能减退症风险降低相关,为药物再利用提供了潜在靶点。
文章的局限:
- 研究主要基于欧洲人群,其结果在其他祖先群体中的普适性仍需验证。
- 甲状腺功能减退症的表型定义依赖于自我报告或汇总统计数据,可能引入异质性。
- PRS在预测甲状腺功能减退症方面仍需与临床指标结合,才能达到最佳预测效果。
- 研究中使用的炎症标志物面板可能不全面,无法完全捕捉所有相关的炎症途径。
- 尽管识别出潜在致病基因,但其具体功能机制仍需进一步实验验证。
文章标题: Multi-ancestry genome-wide association analyses of polycystic ovary syndrome
中文标题: PCOS多祖先GWAS:揭示多囊卵巢综合征遗传奥秘,指明精准治疗新方向!
关键词: 多囊卵巢综合征、全基因组关联研究、多祖先分析、MYB基因、药物发现
摘要总结:
多囊卵巢综合征(PCOS)是育龄期女性最常见的内分泌疾病,其高度遗传性但多基因结构仍不甚明了。这篇文章通过对12,419名中国女性PCOS患者和34,235名对照进行全基因组关联研究(GWAS),并结合多达13,773例欧洲病例和411,088例对照的多祖先Meta分析,识别出94个独立的基因座,其中73个是首次报道,这对于深入理解PCOS的分子基础和开发新的精准治疗策略具有重要意义。
研究发现,尽管进化压力不同,中国和欧洲祖先群体在遗传上存在显著重叠。整合功能分析优先识别出调控特定组织基因活性、疾病致病基因(包括抗苗勒管激素AMH)以及涉及配体结合域相互作用和过氧化物酶体增殖物激活受体γ(PPARG)信号通路的生物学通路中的调控变异。颗粒细胞在PCOS发展中被认为尤为重要。基因组驱动的药物发现方法揭示了多个药物靶点和药物再利用机会,例如芳香酶抑制剂(如阿那曲唑)和胰岛素增敏剂(如吡格列酮)的作用得到强化,并发现了新的代谢干预靶点(如甜菜碱)。这些结果不仅加深了对PCOS分子基础的理解,还为个性化治疗策略铺平了道路,对于PCOS的诊断、风险分层和靶向治疗具有重要意义。
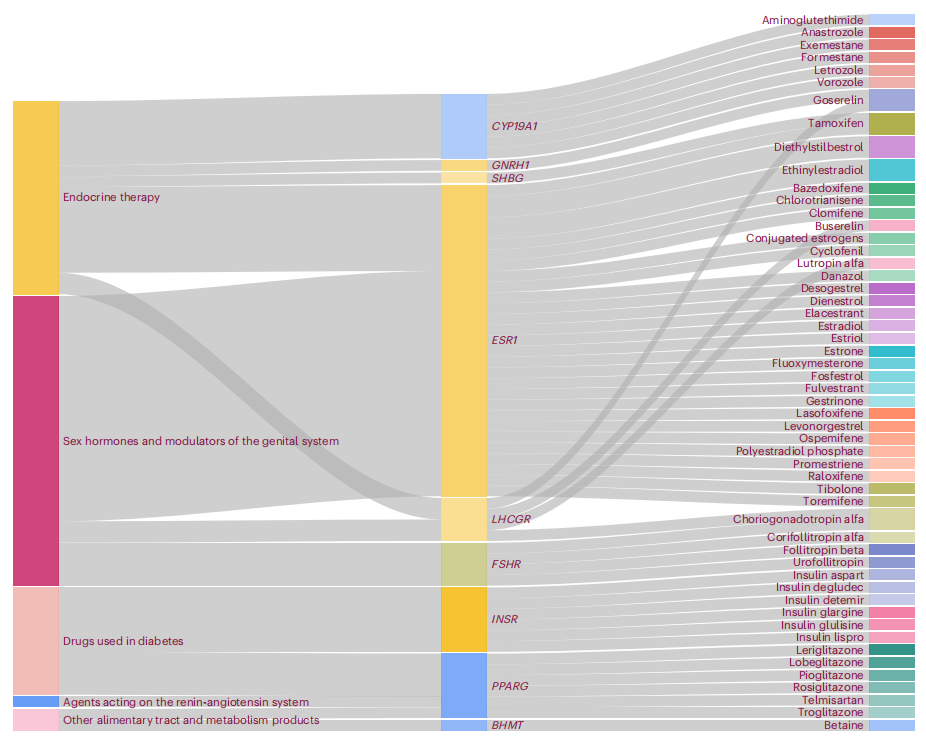
文章的亮点:
- 通过大规模多祖先GWAS识别出94个与PCOS相关的新基因座,其中73个是首次报道。
- 揭示中国和欧洲祖先群体在PCOS遗传上存在显著重叠,尽管进化压力不同。
- 优先识别出调控基因活性、AMH基因以及PPARG信号通路等关键生物学通路中的调控变异。
- 发现颗粒细胞在PCOS发展中具有特别重要的作用。
- 通过基因组驱动的药物发现方法,揭示了多个药物靶点和药物再利用机会,为精准治疗提供了新方向。
文章的局限:
- 部分欧洲数据集依赖自我报告的PCOS诊断,可能引入回忆偏差。
- 缺乏来自东亚和欧洲以外祖先群体的参与者,可能限制结果的普适性。
- 尽管识别出大量相关基因,但未进行全面的功能分析(如细胞或动物实验)。
- 多基因风险评分(PRS)模型在不同祖先群体间的预测准确性存在差异,需要祖先群体特异性校准。
- 研究中使用的基因分型芯片和 imputation 质量差异可能影响结果的准确性。
致谢橙子牛奶糖(陈文燕),请用参考模版:We thank the blogger (orange_milk_sugar, Wenyan Chen) for XXX
感谢小可爱们多年来的陪伴, 我与你们一起成长~

本文来自博客园,作者:橙子牛奶糖(陈文燕),转载请注明原文链接:https://www.cnblogs.com/chenwenyan/p/19287640

 浙公网安备 33010602011771号
浙公网安备 33010602011771号