

Nature Genetics | 本周最新文献速递
文章标题:A pangenome of maize provides genetic insights into drought resistance
中文标题:破译玉米抗旱密码!构建超级泛基因组揭示关键基因
关键词:玉米、泛基因组、干旱胁迫、结构变异、基因功能
摘要总结:干旱是全球农业生产面临的严峻挑战,严重威胁着玉米等主要作物的产量和稳定性。深入解析作物抗旱的遗传基础,对于培育耐旱新品种、保障粮食安全至关重要。这篇文章通过对25个在抗旱性方面表现出显著差异的玉米种质资源进行高质量的全基因组从头组装,并结合已有的31个玉米基因组序列,构建了一个包含56个种质的、迄今为止最全面的玉米泛基因组图谱。基于这一强大的基因组资源,研究人员系统地进行了泛基因组分析、结构变异(SV)鉴定和全基因组关联研究(GWAS),探索了玉米中广泛的遗传变异如何影响其在不同生长阶段(苗期和开花期)的抗旱能力,并深入挖掘了控制这一复杂性状的关键基因和分子机制。研究发现,在脱落酸(ABA)相关或干旱相关基因中存在着丰富的稀有等位基因变异和广泛的调控多样性,这些可能是导致不同种质间抗旱能力差异的重要原因。更重要的是,文章成功鉴定并功能验证了三个新的抗旱关键基因:ZmUGE2、ZmSIL2_和_ZmASI3。功能实验表明,_ZmUGE2_通过加强细胞壁的机械支撑来增强抗旱性;_ZmSIL2_作为一个转录因子,调控胁迫响应基因的表达;而_ZmASI3_则在干旱条件下协调雌雄穗的发育,减少“花期不遇”现象,从而降低产量损失。这对于深入理解植物抗旱的遗传调控网络、为玉米的分子设计育种提供宝贵的基因资源和理论依据,以及最终加速培育抗旱玉米新品种以应对全球气候变化具有重要意义。

文章的亮点:
- 资源构建的全面性:构建了当时最全面的玉米泛基因组(包含56个高质量基因组),极大地扩展了玉米的遗传变异图谱,为深入研究玉米复杂性状提供了前所未有的资源。
- 多阶段抗旱机制解析:研究不仅关注了苗期的抗旱性,还重点解析了对产量影响更为关键的开花期抗旱机制,提供了更完整的抗旱调控网络视图。
- 关键新基因的发现与验证:成功鉴定并系统地验证了三个全新的、在不同层面发挥作用的抗旱基因(ZmUGE2、ZmSIL2、ZmASI3),揭示了细胞壁力学、胁迫信号转导和生殖发育协同调控的新机制。
- 育种应用价值巨大:发现的稀有有利等位变异和关键基因为玉米抗旱育种提供了精确的分子标记和改良靶点,具有很强的实际应用潜力。
文章的局限:
- 群体水平SV基因分型的局限性:在较大的关联群体中对泛基因组的所有结构变异进行精确分型仍然具有挑战性,当前研究主要依赖于短读长测序比对到线性参考基因组,这可能导致对复杂结构变异的检测不准确或遗漏。
- 功能验证的范围有限:尽管文章对三个关键基因进行了深入的功能验证,但泛基因组分析揭示的大量稀有等位变异和调控多样性的具体生物学功能仍有待进一步的实验验证。
- 环境互作的复杂性:研究主要在控制条件下进行,而田间自然干旱环境更为复杂多变。基因型与环境的互作效应可能影响这些基因在不同生态条件下的实际表现,需要更广泛的田间试验。
文章标题:Genetic diversity and evolution of rice centromeres
中文标题:揭秘水稻着丝粒的遗传多样性与进化驱动力!
关键词:水稻、着丝粒、遗传多样性、进化、逆转录转座子
摘要总结:着丝粒是确保染色体在细胞分裂过程中正确分离的关键结构,其动态变化是真核生物进化和物种形成的核心驱动力,但其序列的高度重复性给研究带来了巨大挑战。这篇文章通过对来自Oryza AA基因组群的67个水稻基因组进行组装,并分析了超过800个近乎完整的着丝粒序列,探索了水稻着丝粒的遗传多样性、结构层次和进化机制。研究人员开发了一套新的分析框架,通过从头注释着丝粒卫星序列CEN155,并采用渐进式压缩策略,精确量化了水稻卫星阵列的局部同源化和多层嵌套结构。研究结果表明,水稻着丝粒的遗传创新主要来源于结构变异和“嗜着丝粒”逆转录转座子的插入。与染色体臂区相比,着丝粒区的单碱基替换率相对较低。通过对着丝粒CEN155阵列、逆转录转座子和功能性着丝粒(由CENH3蛋白结合区域定义)的比较分析,揭示了它们之间动态但又相互关联的演化关系。与拟南芥着丝粒进化的KARMA模型不同,该研究提出了一个新的假说:逆转录转座子的入侵可能促进了祖先着丝粒卫星阵列的衰退,并推动了着丝粒的重定位。这一假说得到了CENH3染色质免疫共沉淀测序信号在天然卫星阵列之外区域富集的证据支持。这对于深入理解植物着丝粒的结构复杂性、动态演化规律及其在物种形成中的作用具有重要意义,为研究染色体生物学和进化遗传学提供了新的视角和宝贵的数据资源。
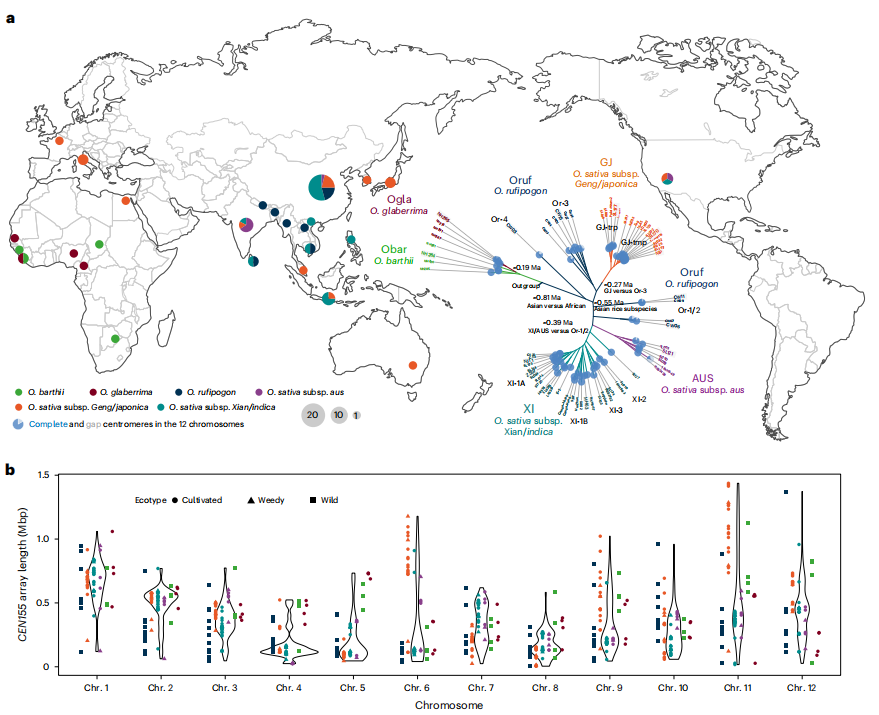
文章的亮点:
- 前所未有的规模和完整性:首次对Oryza AA基因组群的70个高质量基因组进行了近乎完整的着丝粒组装和分析,揭示了前所未有的着丝粒遗传多样性。
- 创新的分析方法:开发并应用了一套新的分析框架,包括渐进式压缩策略,能够有效解析高度重复的着丝粒序列的局部同源化和多层嵌套结构。
- 提出了新的进化模型:基于详实的数据,对经典的着丝粒进化模型提出了挑战,提出了一个由逆转录转座子入侵驱动的“祖先卫星阵列衰退-着丝粒重定位”新假说。
- 遗传与表观遗传的关联:结合了基因组序列分析和CENH3 ChIP-seq数据,关联了着丝粒的序列结构与其功能(动粒附着位点),揭示了遗传与表观遗传在着丝粒进化中的协同作用。
文章的局限:
- 物种范围的局限性:研究主要集中在Oryza AA基因组群,其结论是否能推广到更广泛的植物界,特别是那些具有不同类型着丝粒(如全着丝粒)的物种,尚需进一步研究。
- 进化时间尺度的推断:虽然研究揭示了动态的演化模式,但对于这些变化的精确时间尺度和选择压力等进化参数的推断仍较为初步。
- 功能验证的缺乏:研究提出的关于逆转录转座子功能的假说主要基于相关性分析和计算推断,缺乏直接的实验证据来验证其在着丝粒衰退和重定位中的因果作用。
文章标题:Complete genome assemblies of two mouse subspecies reveal structural diversity of telomeres and centromeres
中文标题:首次实现小鼠端粒到端粒的完整基因组组装
关键词:小鼠基因组、端粒到端粒(T2T)、结构多样性、着丝粒、端粒
摘要总结:小鼠是理解哺乳动物疾病生物学的关键模型,但其参考基因组发布二十多年来仍存在大量缺口,尤其是在端粒和着丝粒等高度重复区域,限制了对这些关键结构域的研究。这篇文章通过采用单分子超长读长测序技术,探索了两个关键近交系小鼠亚种(C57BL/6J和CAST/EiJ)基因组的完整结构,并成功构建了首个小鼠端粒到端粒(T2T)的完整基因组。这些T2T基因组不仅填补了现有参考基因组(GRCm39)中的所有常染色体缺口,还首次完整地呈现了所有常染色体的端粒和着丝粒序列,为参考基因组新增了超过213Mb的新序列和517个新的蛋白质编码基因。研究发现,这两个亚种在端粒和着丝粒的大小及结构组织上表现出显著的变异性。此外,文章还对两个重要但之前不完整的基因座——性染色体上的假常染色体区(PAR)和KRAB锌指蛋白基因座——进行了深入分析。研究揭示了PAR边界在不同品系间的差异、节段性重复的拷贝数和大小变化,以及PAR基因中的大量氨基酸替换突变。这对于全面理解哺乳动物基因组的结构、功能和进化,特别是染色体末端和中心区域的生物学,以及解析与这些区域相关的疾病遗传基础具有重要意义,为未来的功能实验和进化分析提供了前所未有的高质量基因组资源。

文章的亮点:
- 里程碑式的技术突破:首次实现了小鼠这一重要模式生物的端粒到端粒(T2T)完整基因组组装,解决了长期以来困扰基因组学领域的高度重复区域组装难题。
- 揭示了巨大的结构多样性:通过比较两个不同亚种的T2T基因组,揭示了在着丝粒、端粒、假常染色体区(PAR)等关键功能区域存在着惊人的结构变异和序列多样性。
- 填补了基因组的“暗物质”:为小鼠参考基因组增加了大量新序列和数百个新基因,极大地丰富了我们对小鼠基因组内容和功能的认识,为研究这些新发现基因的功能铺平了道路。
- 提供了宝贵的比较基因组学资源:高质量的T2T基因组为研究染色体进化、物种形成、基因组不稳定性以及与重复序列相关的疾病提供了精确的模型和工具。
文章的局限:
- Y染色体的缺失:尽管常染色体和X染色体达到了T2T级别,但Y染色体由于其更加复杂的重复结构,在此次研究中仍未能完全组装,是基因组中尚待完成的部分。
- 品系的局限性:研究仅限于两个特定的近交系(C57BL/6J和CAST/EiJ),虽然它们代表了不同的亚种,但仍无法完全覆盖小鼠物种内广泛的遗传多样性。
- 功能注释的初步性:新发现的517个蛋白质编码基因的功能注释主要基于序列同源性预测,其真实的生物学功能和在生理病理过程中的作用需要大量的后续实验验证。
文章标题:Genotyping sequence-resolved copy number variation using pangenomes reveals paralog-specific global diversity and expression divergence of duplicated genes
中文标题:基于泛基因组解析拷贝数变异的基因分型揭示旁系同源基因特异性的全球多样性及重复基因的表达差异
关键词:拷贝数变异、泛基因组、基因分型、ctyper、旁系同源基因
摘要总结:拷贝数变异(CNV)基因在进化和疾病中扮演着重要角色,但其内部的序列变异在传统大规模研究中仍是一个盲点。这篇文章利用泛基因组资源,开发了一种名为ctyper的新方法,探索了如何从二代测序样本中精确解析CNV基因的等位基因特异性拷贝数和局部单倍型。ctyper方法能够有效地处理包括非参考重复、基因转换和复杂重排在内的复杂变异。在对3,351个CNV基因和212个医学相关的挑战性基因(CMR基因)的基准测试中,ctyper在CNV基因中实现了对96.5%的定相变异的捕获,拷贝数正确率超过99.1%;在CMR基因中,这一比例也达到了94.8%。该方法计算效率高,在单个CPU上仅需1.5小时即可完成一个基因组的分型。应用ctyper的分型结果,研究发现其对基因表达的预测能力相比已知的eQTL变异提升了4.81倍。进一步分析揭示,7.94%的旁系同源基因存在显著的表达分歧,4.68%存在组织特异性的表达偏好。例如,研究发现_SMN1_基因转换为_SMN2_导致_SMN2_表达量降低,这可能影响脊髓性肌萎缩症的病理;同时,_AMY2B_基因的易位重复导致其表达量增加。这对于在生物银行(biobank)规模上实现对CNV和CMR基因的精确分型具有重要意义,为深入理解重复基因的全局多样性、表达分歧及其在复杂疾病中的作用提供了强有力的工具。

文章的亮点:
- 方法学创新:开发了名为ctyper的新型基因分型工具,首次实现了利用泛基因组对复杂CNV区域进行序列解析级别的等位基因特异性拷贝数分型。
- 高精度与高效率:在包括医学相关基因在内的大量复杂基因上展示了极高的准确性(>99%)和检出率(>94%),同时保持了适用于大规模生物银行项目的高计算效率。
- 揭示了新的生物学见解:通过对等位基因特异性表达的量化,揭示了旁系同源基因间广泛存在的表达分歧和组织特异性偏好,并以_SMN1/SMN2_和_AMY2B_为例,展示了其在疾病相关基因中的潜在功能影响。
- 提升了基因表达预测能力:证明了序列解析的CNV分型结果能显著提高对基因表达水平的预测能力,远超传统eQTL变异的效果,强调了精细遗传变异在调控网络中的重要性。
文章的局限:
- 依赖于泛基因组的完整性:ctyper的性能高度依赖于所使用的泛基因组参考数据库的质量和全面性。如果某个样本中存在数据库未包含的稀有或新的单倍型,分型准确性可能会下降。
- 亚群粒度的限制:由于样本量的限制,当前的关联分析主要基于较大的“亚群”(subgroups)而非单个等位基因(PAs),这可能掩盖了更精细等位基因之间的功能差异。
- 短读长测序的固有局限:尽管该方法针对二代测序数据进行了优化,但在极度复杂或超长重复区域,短读长数据本身的信息含量仍然有限,可能导致分型模糊或错误。
- 表达分析的局限:eQTL分析仅限于能够被RNA-seq明确映射的基因,对于那些转录本高度相似以至于无法区分的旁系同源基因,其表达量只能合并分析,限制了对个体旁系基因表达调控的解析。
文章标题:Locityper enables targeted genotyping of complex polymorphic genes
中文标题:Locityper实现对复杂多态性基因的精准靶向分型
关键词:基因分型、多态性基因、泛基因组、Locityper、结构变异
摘要总结:人类基因组中包含数百个与疾病相关的、结构高度可变的多态性基因座,这些区域因其复杂性而难以通过标准方法进行准确的变异检测。这篇文章通过开发一种名为Locityper的新工具,探索了如何利用短读长和长读长全基因组测序数据,对这些具有挑战性的基因进行靶向分型。Locityper首先从泛基因组等资源中提取目标基因座的已知单倍型,然后招募并比对测序读长至这些单倍型上,通过优化读长比对、插入片段大小和读长深度分布等多个维度的信息,找到最可能的单倍型组合。在对256个医学相关的挑战性基因座的测试中,无论是使用短读长还是长读长数据,Locityper的分型质量值(QV)中位数均超过35,其性能分别比当前顶尖的Illumina和PacBio HiFi变异检测流程高出10.9和1.7个点。此外,Locityper还能有效处理如HLA、KIR、MUC和FCGR等超多态性基因家族。由于其高效的计算性能,Locityper能够扩展到生物银行规模的队列研究中。这对于开启对以往难以分析的疾病相关基因的关联研究具有重要意义,为揭示复杂疾病的遗传基础提供了强大的新工具。

文章的亮点:
- 技术创新性:提出了一种新的靶向基因分型策略,整合了读长比对、插入片段大小和深度等多源信号,专门用于解析传统方法难以处理的高度多态和结构复杂的基因座。
- 高性能与广适用性:在短读长和长读长数据上均表现出超越现有主流变异检测流程的卓越准确性,证明了其在不同测序平台上的强大适用性。
- 攻克关键医学基因:成功应用于HLA、KIR、MUC等多个以极度多态性著称的、与免疫和多种疾病密切相关的基因家族,展示了其在医学研究中的巨大潜力。
- 可扩展性强:算法计算效率高,使其能够轻松扩展到数万甚至数十万样本的生物银行级别队列,为这些大规模数据集中“不可见”的遗传变异研究铺平了道路。
文章的局限:
- 依赖于参考单倍型库:Locityper的准确性依赖于一个全面的单倍型参考面板。如果一个样本携带的单倍型在参考库中不存在或代表性不足,分型准确性会显著下降。
- 无法发现新单倍型:该工具的设计目标是“识别”已知的单倍型,而不是“发现”新的单倍型。它只能将样本的序列匹配到参考库中最相似的单倍型,而不能从头构建新的等位基因。
- 对旁系同源基因的区分能力有限:当两个或多个高度同源的基因座(旁系同源基因)在基因组中物理位置接近时,可能会存在读长招募的交叉干扰,影响分型的准确性。
- 在极长或高度重复区域的挑战:尽管性能优越,但在处理含有极长串联重复(如某些MUC基因的VNTR区域)的基因座时,短读长数据仍然存在固有的局限性,可能导致分型精度下降。
文章标题:Real-time dynamic polygenic prediction for streaming data
中文标题:实时动态多基因风险预测模型(rtPRS-CS)问世
关键词:多基因风险评分、精准医疗、流式数据、动态预测、rtPRS-CS
摘要总结:多基因风险评分(PRS)是推动精准医疗的重要工具,但现有方法依赖于静态的全基因组关联研究(GWAS)摘要统计数据,更新周期长,无法充分利用医疗保健领域持续产生的新基因和健康数据。这篇文章通过在PRS-CS框架基础上进行扩展,开发了一种名为实时PRS-CS(rtPRS-CS)的新方法,探索了如何随着每个新样本的收集,在线、动态地优化和标准化PRS,从而实现对后续患者风险的更精准预测。rtPRS-CS利用随机梯度下降算法,在每个新样本加入时迭代更新SNP权重,整个过程计算高效。研究通过广泛的模拟评估了rtPRS-CS在不同遗传结构和训练样本量下的性能。利用两个大规模生物银行(MGBB和UKBB)的定量性状数据,研究表明rtPRS-CS能够整合海量流式数据,随时间推移显著提升PRS的预测能力。此外,研究还将rtPRS-CS应用于包含七个亚洲地区的22个精神分裂症队列,证明了该方法在动态捕捉不同遗传背景人群的健康状况变化和预测疾病风险方面的临床效用。这对于在真实世界的医疗环境中,最大化利用持续增长的数据以提供最准确的遗传风险预测具有重要意义,推动了PRS从静态研究工具向动态临床决策支持工具的转变。

文章的亮点:
- 方法学上的范式转变:首次提出并实现了一个实时、动态更新的多基因风险评分(PRS)框架,打破了传统PRS依赖静态、周期性更新的GWAS摘要统计的局限。
- 高效的在线学习算法:巧妙地运用随机梯度下降算法,使得模型可以在每个新样本加入时进行快速、轻量级的权重更新,极大地提高了计算效率和应用的时效性。
- 跨人群和跨性状的稳健性:通过在模拟数据、两个大型生物银行的多种定量性状以及跨越多个亚洲人群的精神分裂症队列中的成功应用,证明了该方法的强大稳健性和广泛适用性。
- 动态适应真实世界场景:模型不仅能整合新样本以提高预测准确性,还能动态调整和标准化PRS,以适应人群结构变化和个体健康状态的动态变化(如从健康对照转为病例),更贴近复杂的临床现实。
文章的局限:
- 对初始模型参数的依赖:rtPRS-CS的性能在一定程度上依赖于初始基线GWAS提供的全局和局部遗传度参数。如果初始参数估计不准(尤其是在小样本基线研究中),可能会影响后续更新的效率和最终的准确性。
- 样本顺序和相关性的潜在影响:尽管研究表明最终结果与样本顺序无关,但在训练过程中,样本的加入顺序以及样本间的亲缘关系可能导致预测评分的短期波动,这在临床应用中需要谨慎处理。
- 对复杂遗传结构的扩展性:当前模型主要适用于相对同源的欧洲人群,虽然在亚洲人群中也表现良好,但对于高度混合或跨大陆人群的直接应用仍具挑战,需要与PRS-CSx等跨人群方法进一步整合。
- 无法替代周期性GWAS:该方法是一种高效的“增量更新”工具,但不能完全替代对表型和基因型数据进行严格质控和协调的周期性大规模GWAS,后者对于校正批次效应和获得最精确的效应量估计仍然至关重要。
文章标题:An African ancestry-specific nonsense variant in CD36 is associated with a higher risk of dilated cardiomyopathy
中文标题:非洲裔人群扩张性心肌病高风险之谜揭晓:CD36基因变异是关键
关键词:扩张性心肌病、非洲裔、CD36、全基因组关联研究、功能丧失变异
摘要总结:扩张性心肌病(DCM)在非洲裔人群中负担尤为严重,其背后的遗传原因尚不完全清楚。这篇文章通过对包含1,802名非洲裔DCM病例和93,804名对照的大规模全基因组关联研究(GWAS),探索了导致非洲裔人群DCM风险升高的特异性遗传因素。研究发现,_CD36_基因中的一个无义变异(rs3211938:G )与DCM风险显著增加相关。该变异在非洲裔人群中频率较高(约17%),但在欧洲裔人群中几乎不存在(<0.1%),这可能与它在疟疾抵抗中提供的保护性选择优势有关。携带该风险等位基因纯合子的个体(约占非洲裔人群的1%),其DCM风险增加了约三倍。在没有临床心肌病的个体中,纯合子也表现出左心室射血分数的显著降低,呈现亚临床表型。该单一变异对非洲裔人群DCM的群体归因分数达到8.1%,解释了与欧洲裔相比约20%的额外DCM风险。进一步的实验验证,利用人诱导多能干细胞来源的心肌细胞模型证明,_CD36_基因的功能丧失会损害心肌细胞对脂肪酸的摄取,破坏心脏的能量代谢和收缩功能。这对于揭示非洲裔人群中DCM的一个重要且普遍的遗传病因,阐明心肌能量代谢受损作为DCM发病机制的核心环节,以及为针对特定人群的DCM风险筛查和潜在治疗干预具有重要意义。

文章的亮点:
- 重大发现:首次发现了一个在非洲裔人群中常见、但在其他人群中罕见的_CD36_无义变异是DCM的主要风险因素,为解释该人群中的健康不平等现象提供了重要的遗传学证据。
- 清晰的机制阐释:通过“GWAS-精细定位-功能实验”的完整研究链,清晰地证明了_CD36_功能丧失导致心肌脂肪酸代谢障碍,进而引发收缩功能障碍,最终导致DCM的病理生理通路。
- 进化与疾病的关联:将该风险变异与疟疾抵抗的阳性选择压力联系起来,为“进化权衡”(evolutionary trade-off)理论提供了一个生动的实例,即一个在特定环境下有利的基因变异可能在另一环境下导致疾病风险。
- 临床转化潜力:研究结果提示_CD36_变异可作为非洲裔人群DCM风险分层的重要生物标志物,并且为开发靶向心肌能量代谢的治疗策略提供了新的思路。
文章的局限:
- 病例定义的局限性:DCM病例的确定主要基于电子健康记录中的诊断代码,可能存在诊断不精确或分类错误的情况,但这通常会使关联结果偏向于无效假设,从而低估了真实的关联强度。
- 未考虑其他遗传因素:在评估该变异对人群风险差异的贡献时,研究未全面考虑其他可能存在的、在不同人群中频率各异的DCM风险或保护性遗传变异。
- 功能实验模型的简化:体外hiPSC心肌细胞模型虽然揭示了核心分子机制,但它使用的是siRNA介导的完全敲低,而非直接引入rs3211938变异,且无法完全模拟体内复杂的生理环境和长期的代偿与失代偿过程。
- 成像队列样本量较小:用于验证亚临床表型的多中心影像学队列中,携带纯合风险变异的个体数量较少,尽管结果一致且统计学显著,但更大的样本量将能提供更稳健的效应量估计。
致谢橙子牛奶糖(陈文燕),请用参考模版:We thank the blogger (orange_milk_sugar, Wenyan Chen) for XXX
感谢小可爱们多年来的陪伴, 我与你们一起成长~

本文来自博客园,作者:橙子牛奶糖(陈文燕),转载请注明原文链接:https://www.cnblogs.com/chenwenyan/p/19191259

 浙公网安备 33010602011771号
浙公网安备 33010602011771号