FEM:整合RANSEQ和DNA甲基化数据分析的R包
FEM是一个整合RANSEQ和DNA甲基化数据的R包,由Andrew E. Teschendorff 和 Zhen Yang 开发、维护。
不多说,下面介绍如何使用FEM整合RANSEQ和DNA甲基化数据分析。
1、安装、下载FEM
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("FEM")
library(FEM)
2、下载数据adj.m
adj.m数据存储在网址(https://sourceforge.net/projects/signalentropy/files/?source=navbar)的“hprdAsigH-13Jun12.Rd”上,下载“hprdAsigH-13Jun12.Rd”即可。
“hprdAsigH-13Jun12.Rd”文件包含三个数据:"hprdAsigH.m"、"sigHclassA.v"、"sigHclassA2.v"
"hprdAsigH.m"为我们后续分析需要的数据。
“hprdAsigH-13Jun12.Rd”数据也可以通过公众号bio生物信息后台发生关键字“FEM”获得。
3、准备DNA甲基化数据
DNA甲基化数据取得是beta值,这里我们命名为“beta”,示例图如下所示:

行名为每个CpG位点的ID,列名为每个样本的ID。
4、准备DNA甲基化数据对应的表型文件
DNA甲基化数据对于的表型文件,我们命名为group,其示例图如下所示:

表示为第一个样本sample1是control,第二个样本sample2是control,第三个样本sample3是case,以此类推。beta文件的sample和group是一一对应的。
5、准备RANSEQ基因表达数据
RANSEQ基因表达数据,我们命名为need,其示例如下所示:

行名是每一个基因的entrez gene IDs,列名是每一个样本名。
6、准备RANSEQ基因表达数据对应的表型文件
RANSEQ基因表达数据对应的表型文件,我们命名为gg, 示例图如下所示:
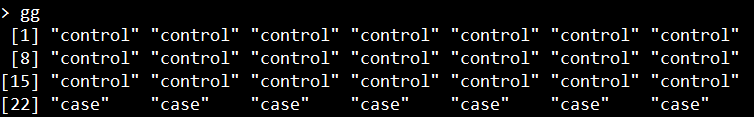
表示的是每一个样本对应的是case还是control。与DNA甲基化的情况一样,need文件的sample和group是一一对应的。
7、生成DNA差异甲基化统计量
如果是850k,则用以下命令:
statM.o=GenStatM(beta,group,"EPIC")
如果是450K,则用以下命令:
statM.o=GenStatM(beta,group,"450K")
生成的statM.o结果包含三个数据:"top"、 "cont"、"avbeta"
"top"是差异甲基化的统计结果,top是一个list,差异甲基化结果一般存储在top的第一个元素(item)中;
"cont"是差异甲基化分析时构建的case-control;
"avbeta"是DNA甲基化数据;
8、生成差异表达的统计量
使用命令:
statR.o=GenStatR(need,gg)
生成的statR.o结果包含三个数据: "top"、"cont"、"avexp"
与差异甲基化的结果类似, "top"是差异表达的统计结果;
"cont"是差异表达分析时构建的case-control;
"avbeta"是表达数据;
9、整合差异表达和差异甲基化数据
load("/data/chenwenyan/hprdAsigH-13Jun12.Rd")
re=DoIntFEM450k(statM.o,statR.o,hprdAsigH.m,1,1,"avbeta")
解释一下,statM.o和statR.o分别是步骤7和8产生的结果文件,hprdAsigH.m是步骤2下载的“hprdAsigH-13Jun12.Rd”文件包含的数据,这里我存储在/data/chenwenyan/路径下,请读者们根据各自存储的路径自行修改,不要完成照抄我的路径。
两个1分别指的是statM.o和statR.o的top数据的第一个文件,即步骤7和8生成的差异甲基化和差异表达结果。
这里需要注意的是,如果你感兴趣的分组结果存储在top数据的第二个元素,则代码需要改成re=DoIntFEM450k(statM.o,statR.o,hprdAsigH.m,2,2,"avbeta")
10、鉴定甲基化与表达之间存在负相关的基因
DoFEMbi=DoFEMbi(re, nseeds = 100, gamma = 0.5, nMC = 1000, sizeR.v = c(1,100), minsizeOUT = 10, writeOUT = TRUE, nameSTUDY = "TEST", ew.v = NULL)
这里所有参数均可以使用默认值。
输出的DoFEMbi结果包含以下文件:

这里我们主要关注fem和topmod这两个元素,分别指的是模块以及模块对应的统计数据,如下所示:

11、可视化结果
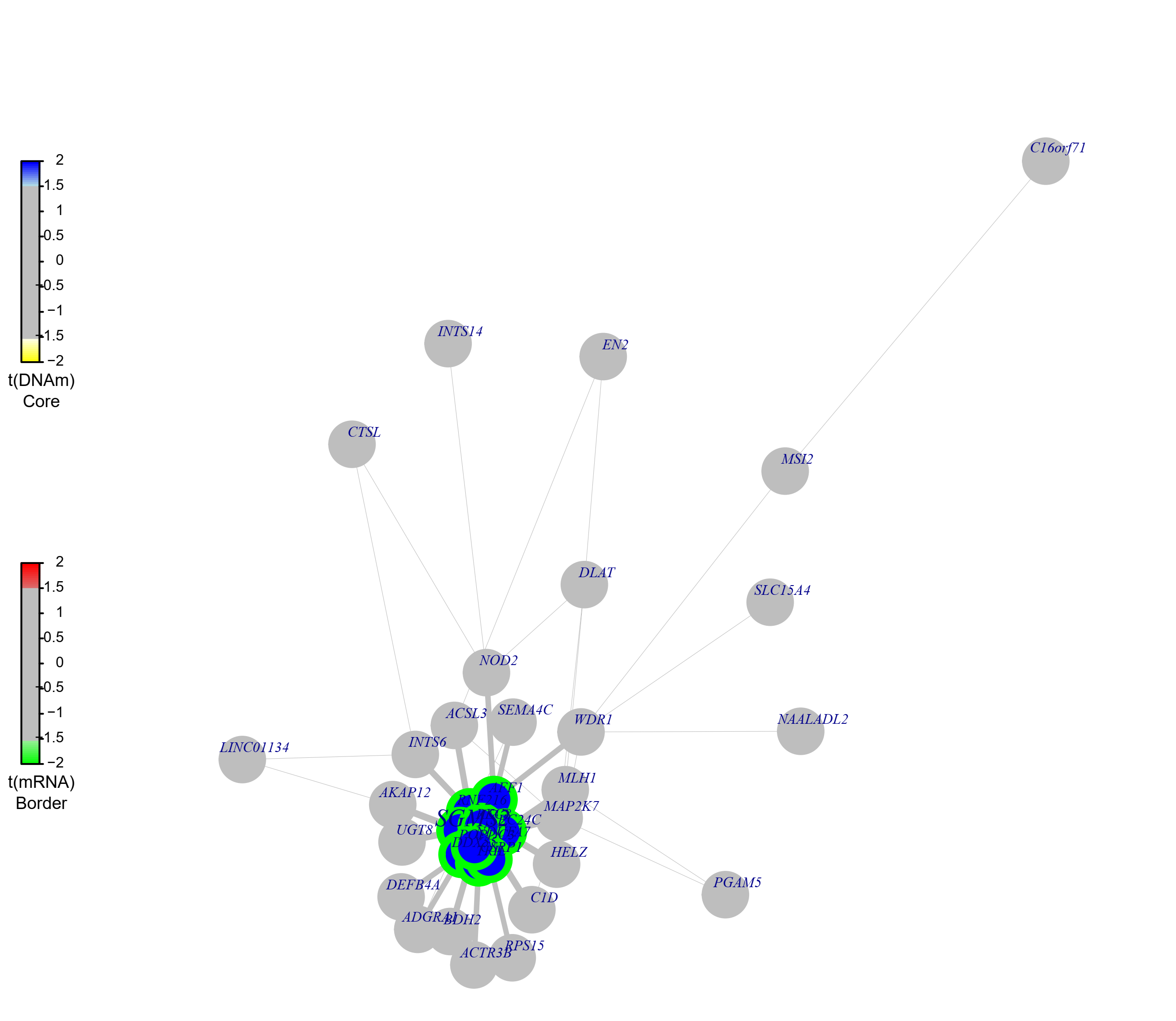
可视化SGMS2模块信息:
SGMS2=FemModShow(DoFEMbi$topmod$SGMS2,name="SGMS2", DoFEMbi)
画出来的图如下所示,可以看到,这个模块的基因主要是高甲基化低表达:

本文来自博客园,作者:橙子牛奶糖(陈文燕),转载请注明原文链接:https://www.cnblogs.com/chenwenyan/p/13658704.html

 浙公网安备 33010602011771号
浙公网安备 33010602011771号