

ATAC-seq数据分析
参考文献:
Cohen, Lianne B et al. “A genome-wide survey reveals a diverse array of enhancers coordinate the Drosophila innate immune response.” bioRxiv : the preprint server for biology 2025.09.24.678314. 24 Sep. 2025, doi:10.1101/2025.09.24.678314. Preprint.
数据准备
- 测序数据
- 参考基因组
- 基因注释
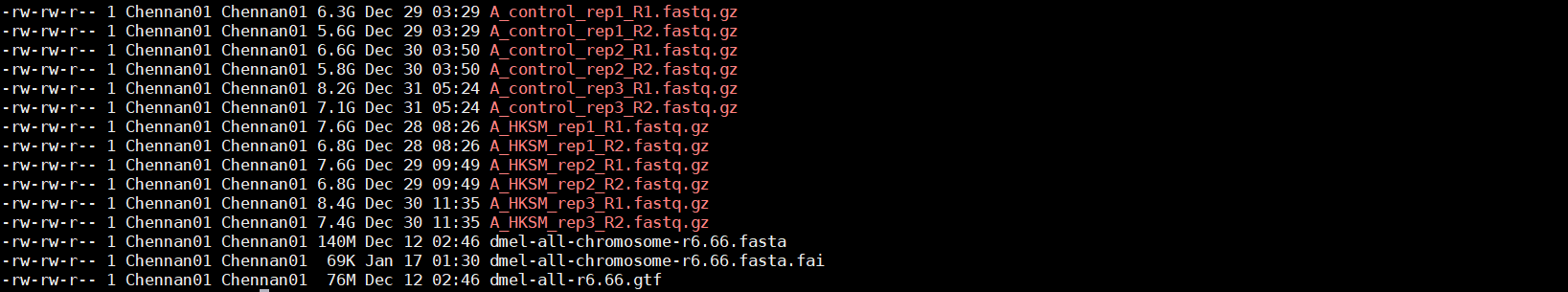
- 样本名称表。rawdata/ATCTseq_list.txt

# 生成样本信息,第一行样本名,第二行样本存储路径
ls $(pwd)/A_* >ATCTseq_list.txt && \
awk -F'/' '{sample=$NF; gsub(/\.fastq\.gz$/, "", sample); print sample "\t" $0}' ATCTseq_list.txt >tmp.txt && \
mv tmp.txt ATCTseq_list.txt1.测序数据质控:fastp
(1)质控(fastp)
fastp 示例
fastp \
-i rawdata/ZAT6_ABA_rep2_R1.fastq.gz \ # read1
-o cleandata/ZAT6_ABA_rep2_R1.fastq.gz \
-I rawdata/ZAT6_ABA_rep2_R1.fastq.gz \ # read2
-O cleandata/ZAT6_ABA_rep2_R1.fastq.gz \
--html cleandata/ZAT6_ABA_rep2_fastp.html \ # 输出报告
--json cleandata/ZAT6_ABA_rep2_fastp.json \
--thread 1 \
--adapter_sequence AGATCGGAAGAGCACACGTCTGAACTCCAGTCA \ # 接头序列,可不设,会自己检测。默认启动接头处理
--adapter_sequence_r2 AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT \
--qualified_quality_phred 20 \ # Q20
--length_required 50 \ # reads<50丢弃
&>logs/ZAT6_ABA_rep2_fastp_quality_control.log生成脚本
cd /data1/home/Chennan01/RNA-Seq/starrseq/cleandata
awk '{print "fastp \
-i ../rawdata/"$1"_R1.fastq.gz \
-o ./"$1"_R1.fastq.gz \
-I ../rawdata/"$1"_R2.fastq.gz \
-O ./"$1"_R2.fastq.gz \
--html ./"$1"_fastp.html \
--json ./"$1"_fastp.json \
--thread 8 \
--qualified_quality_phred 20 \
--length_required 50 \
&>../logs/"$1"_fastp_quality_control.log"}' ../rawdata/ATAC_samplename.txtpbs脚本
(6个任务,分两次同时提交,一次3个任务、8线程)
#!/bin/bash
#PBS -q large_q
#PBS -l nodes=1:ppn=8
#PBS -l mem=64gb
cd /data1/home/Chennan01/RNA-Seq/starrseq/cleandata
fastp -i ../rawdata/A_control_rep1_R1.fastq.gz -o ./A_control_rep1_R1.fastq.gz -I ../rawdata/A_control_rep1_R2.fastq.gz -O ./A_control_rep1_R2.fastq.gz --html ./A_control_rep1_fastp.html --json ./A_control_rep1_fastp.json --thread 8 --qualified_quality_phred 20 --length_required 50 &>../logs/A_control_rep1_fastp_quality_control.log
fastp -i ../rawdata/A_control_rep2_R1.fastq.gz -o ./A_control_rep2_R1.fastq.gz -I ../rawdata/A_control_rep2_R2.fastq.gz -O ./A_control_rep2_R2.fastq.gz --html ./A_control_rep2_fastp.html --json ./A_control_rep2_fastp.json --thread 8 --qualified_quality_phred 20 --length_required 50 &>../logs/A_control_rep2_fastp_quality_control.log
fastp -i ../rawdata/A_control_rep3_R1.fastq.gz -o ./A_control_rep3_R1.fastq.gz -I ../rawdata/A_control_rep3_R2.fastq.gz -O ./A_control_rep3_R2.fastq.gz --html ./A_control_rep3_fastp.html --json ./A_control_rep3_fastp.json --thread 8 --qualified_quality_phred 20 --length_required 50 &>../logs/A_control_rep3_fastp_quality_control.log
#!/bin/bash
#PBS -q large_q
#PBS -l nodes=1:ppn=8
#PBS -l mem=64gb
cd /data1/home/Chennan01/RNA-Seq/starrseq/cleandata
fastp -i ../rawdata/A_HKSM_rep1_R1.fastq.gz -o ./A_HKSM_rep1_R1.fastq.gz -I ../rawdata/A_HKSM_rep1_R2.fastq.gz -O ./A_HKSM_rep1_R2.fastq.gz --html ./A_HKSM_rep1_fastp.html --json ./A_HKSM_rep1_fastp.json --thread 8 --qualified_quality_phred 20 --length_required 50 &>../logs/A_HKSM_rep1_fastp_quality_control.log
fastp -i ../rawdata/A_HKSM_rep2_R1.fastq.gz -o ./A_HKSM_rep2_R1.fastq.gz -I ../rawdata/A_HKSM_rep2_R2.fastq.gz -O ./A_HKSM_rep2_R2.fastq.gz --html ./A_HKSM_rep2_fastp.html --json ./A_HKSM_rep2_fastp.json --thread 8 --qualified_quality_phred 20 --length_required 50 &>../logs/A_HKSM_rep2_fastp_quality_control.log
fastp -i ../rawdata/A_HKSM_rep3_R1.fastq.gz -o ./A_HKSM_rep3_R1.fastq.gz -I ../rawdata/A_HKSM_rep3_R2.fastq.gz -O ./A_HKSM_rep3_R2.fastq.gz --html ./A_HKSM_rep3_fastp.html --json ./A_HKSM_rep3_fastp.json --thread 8 --qualified_quality_phred 20 --length_required 50 &>../logs/A_HKSM_rep3_fastp_quality_control.log(2)合并为质控报告(combine_fastp_report.R)
Rscript scripts/combine_fastp_report.R \
--input *.json \
--output ./ \ # 不指定则在当前目录建立report文件夹,并保存fastp_summary.RData、fastp_summary.tsv
&>1.log
Rscript scripts/combine_fastp_report.R \
--input *.json \
&>2.log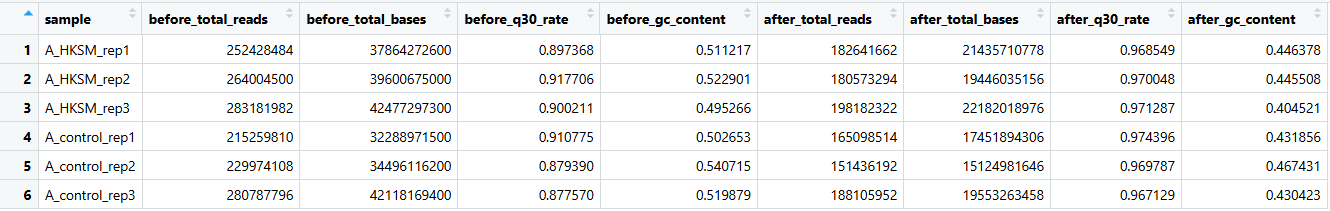
(3)结果
2.比对到参考基因组:bowtie2-build、bowtie2
(1)构建索引:bowtie2-build
bowtie2-build 示例
bowtie2-build dmel-all-chromosome-r6.66.fasta genome # 构建的基因组名称前缀pbs脚本
#!/bin/bash
#PBS -q small_q
#PBS -l nodes=1:ppn=4
#PBS -l mem=32gb
cd /data1/home/Chennan01/RNA-Seq/starrseq/ref
bowtie2-build dmel-all-chromosome-r6.66.fasta genome &>1.log(2)比对:bowtie2
bowtie2 示例
bowtie2 \
-p 1 \ # 线程数1
-x ref/genome \ # 参考基因组索引文件前缀(上面构建好的)
-1 cleandata/Diabetic_hs_rep1_R1.fastq.gz \ # read1
-2 cleandata/Diabetic_hs_rep1_R2.fastq.gz \ # read2
-S raw_bams/Diabetic_hs_rep1.sam \ # 输出sam文件
--no-mixed \ # 不报告混合比对。成对reads都能比对时才报告【ATAC-seq 一般是 PE 测序,设置这两个参数】
--no-discordant \ # 不报告不一致比对。两个reads比对到不同染色体,或方向不符合预期
--maxins 1000 \ # 最大插入片段1000bp,超过过滤。插入片段 = R2 比对结束位置 - R1 比对开始位置
&>logs/Diabetic_hs_rep1_bowtie2_align.log生成脚本
cd /data1/home/Chennan01/RNA-Seq/starrseq/rawbams
awk '{print "bowtie2 -p 10 \
-x ../ref/genome \
-1 ../cleandata/"$1"_R1.fastq.gz \
-2 ../cleandata/"$1"_R2.fastq.gz \
-S ./"$1".sam \
--no-mixed --no-discordant --maxins 1000 \
&>"$1"_align.log"}' ../rawdata/ATAC_samplename.txtpbs脚本
(6个任务,分两次同时提交,一次3个任务、10线程)
#!/bin/bash
#PBS -q large_q
#PBS -l nodes=1:ppn=10
#PBS -l mem=64gb
cd /data1/home/Chennan01/RNA-Seq/starrseq/rawbams
bowtie2 -p 10 -x ../ref/genome -1 ../cleandata/A_control_rep1_R1.fastq.gz -2 ../cleandata/A_control_rep1_R2.fastq.gz -S ./A_control_rep1.sam --no-mixed --no-discordant --maxins 1000 &>A_control_rep1_align.log
bowtie2 -p 10 -x ../ref/genome -1 ../cleandata/A_control_rep2_R1.fastq.gz -2 ../cleandata/A_control_rep2_R2.fastq.gz -S ./A_control_rep2.sam --no-mixed --no-discordant --maxins 1000 &>A_control_rep2_align.log
bowtie2 -p 10 -x ../ref/genome -1 ../cleandata/A_control_rep3_R1.fastq.gz -2 ../cleandata/A_control_rep3_R2.fastq.gz -S ./A_control_rep3.sam --no-mixed --no-discordant --maxins 1000 &>A_control_rep3_align.log
#!/bin/bash
#PBS -q large_q
#PBS -l nodes=1:ppn=10
#PBS -l mem=64gb
cd /data1/home/Chennan01/RNA-Seq/starrseq/rawbams
bowtie2 -p 10 -x ../ref/genome -1 ../cleandata/A_HKSM_rep1_R1.fastq.gz -2 ../cleandata/A_HKSM_rep1_R2.fastq.gz -S ./A_HKSM_rep1.sam --no-mixed --no-discordant --maxins 1000 &>A_HKSM_rep1_align.log
bowtie2 -p 10 -x ../ref/genome -1 ../cleandata/A_HKSM_rep2_R1.fastq.gz -2 ../cleandata/A_HKSM_rep2_R2.fastq.gz -S ./A_HKSM_rep2.sam --no-mixed --no-discordant --maxins 1000 &>A_HKSM_rep2_align.log
bowtie2 -p 10 -x ../ref/genome -1 ../cleandata/A_HKSM_rep3_R1.fastq.gz -2 ../cleandata/A_HKSM_rep3_R2.fastq.gz -S ./A_HKSM_rep3.sam --no-mixed --no-discordant --maxins 1000 &>A_HKSM_rep3_align.log结果

3.排序并转换为bam:samtools
samtools sort 示例
samtools sort \
-@ 1 \ # 1个线程
-o raw_bams/ZAT6_ABA_rep1_sorted.bam \ # 输出
raw_bams/ZAT6_ABA_rep1.sam \ # 输入
&>logs/ZAT6_ABA_rep1_sort_bam.log
samtools index raw_bams/ZAT6_ABA_rep1_sorted.bam生成脚本
cd /data1/home/Chennan01/RNA-Seq/starrseq/rawbams
awk '{print "samtools sort \
-@ 4 \
-o ./"$1"_sorted.bam \
./"$1".sam \
&>"$1"_sort_bam.log && samtools index "$1"_sorted.bam &>>"$1"_sort_bam.log"}' ../rawdata/ATAC_samplename.txtpbs脚本
文件大运行时间长,可分成几个任务一起提交
#!/bin/bash
#PBS -q large_q
#PBS -l nodes=1:ppn=4
#PBS -l mem=32gb
cd /data1/home/Chennan01/RNA-Seq/starrseq/rawbams
samtools sort -@ 4 -o ./A_control_rep1_sorted.bam ./A_control_rep1.sam &>A_control_rep1_sort_bam.log && samtools index A_control_rep1_sorted.bam &>>A_control_rep1_sort_bam.log
samtools sort -@ 4 -o ./A_control_rep2_sorted.bam ./A_control_rep2.sam &>A_control_rep2_sort_bam.log && samtools index A_control_rep2_sorted.bam &>>A_control_rep2_sort_bam.log
samtools sort -@ 4 -o ./A_control_rep3_sorted.bam ./A_control_rep3.sam &>A_control_rep3_sort_bam.log && samtools index A_control_rep3_sorted.bam &>>A_control_rep3_sort_bam.log
samtools sort -@ 4 -o ./A_HKSM_rep1_sorted.bam ./A_HKSM_rep1.sam &>A_HKSM_rep1_sort_bam.log && samtools index A_HKSM_rep1_sorted.bam &>>A_HKSM_rep1_sort_bam.log
samtools sort -@ 4 -o ./A_HKSM_rep2_sorted.bam ./A_HKSM_rep2.sam &>A_HKSM_rep2_sort_bam.log && samtools index A_HKSM_rep2_sorted.bam &>>A_HKSM_rep2_sort_bam.log
samtools sort -@ 4 -o ./A_HKSM_rep3_sorted.bam ./A_HKSM_rep3.sam &>A_HKSM_rep3_sort_bam.log && samtools index A_HKSM_rep3_sorted.bam &>>A_HKSM_rep3_sort_bam.log结果
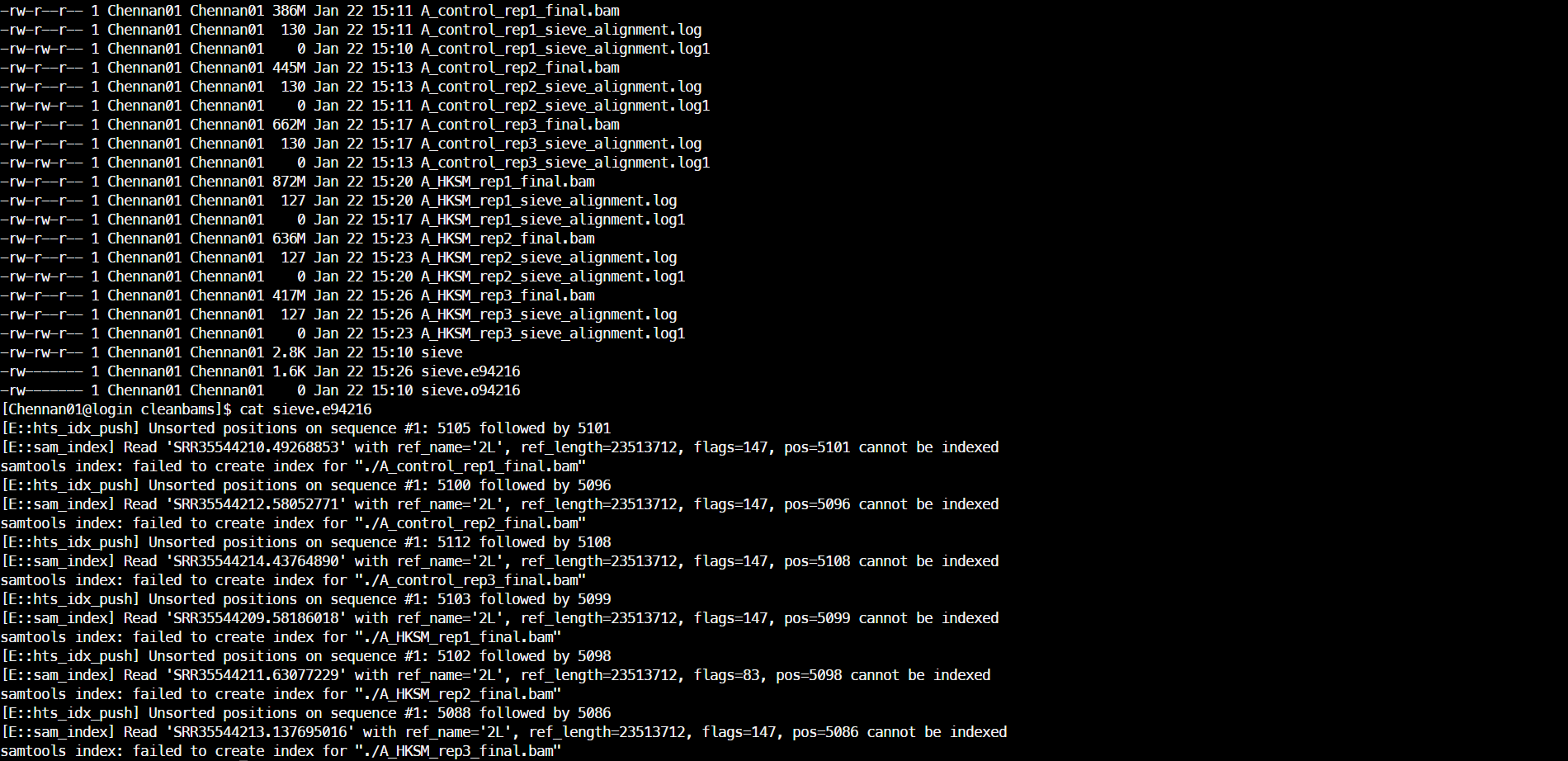
4.比对结果质控:过滤低质量比对、Tn5校正(deeptool alignmentSieve)
(1)过滤
通常的做法是使用 picard 标记 duplicates,然后使用 samtools 去除各种低质量比对。deeptools 包中的 alignmentSieve 能够一步完成这些操作,而且速度更快
这一步过滤
-
duplicates : 经过比较与 Picard 结果差异很小
-
低质量比对:MAPQ 小于 20 或 30
-
未比对上的 reads:
-
SAM flag = 4(reads unmapped)。未成功比对
-
SAM flag = 256(not primary alignment)。次要比对。一个reads比对到两个地方,过滤掉次级比对
-
-
补充比对:SAM flag = 2048(supplementary alignment)
-
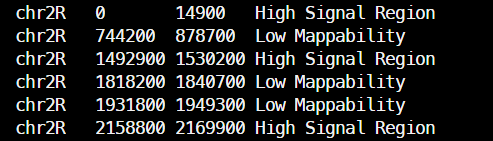
比对到黑名单区域的 reads:黑名单、线粒体(0-全长)、叶绿体、大肠杆菌等
![image]()
# 若无黑名单,对基因组构建索引,生成genome.fa.fai。第一列是染色体名称,第二列是长度 samtools faidx genome.fa![image]()

alignmentSieve 示例
alignmentSieve \
--numberOfProcessors 1 \ # # 并行处理线程数1
--bam raw_bams/ZAT6_ABA_rep1_sorted.bam \ # 输入
--outFile clean_bams/ZAT6_ABA_rep1_final.bam \ # 输出
--filterMetrics logs/ZAT6_ABA_rep1_sieve_alignment.log \ # 过滤结果统计文件
--ignoreDuplicates \ # 去除重复 reads
--minMappingQuality 25 \ # 最小映射质量(MAPQ)阈值,通常范围 0-60,值越高比对越可靠,MAPQ ≥ 25 的 reads 被保留,25 对应 ~0.3% 的错误率
--samFlagExclude 260 \ # 排除特定 SAM flag,260=256+4:排除非主要比对、未比对成功的 reads
--blackListFileName ref/blacklist.bed \ # 指定黑名单区域文件。如端粒、着丝粒等高重复序列,比对质量差的区域、已知的技术假阳性区域
--ATACshift \ # Tn5偏移校正
&>logs/ZAT6_ABA_rep1_sieve_alignment.log # 前面指定后,日志文件无输出,可删
samtools index clean_bams/ZAT6_ABA_rep1_final.bam # 重新排序才能建立索引生成脚本
cd /data1/home/Chennan01/RNA-Seq/starrseq/cleanbams
awk '{print "/usr/local/bin/singularity exec ../sifs/deeptools_20231220.sif alignmentSieve \
--numberOfProcessors 6 \
--bam ../rawbams/"$1"_sorted.bam \
--outFile ./"$1"_final.bam \
--filterMetrics "$1"_sieve_alignment.log \
--ignoreDuplicates \
--minMappingQuality 25 \
--samFlagExclude 260 \
--ATACshift \
--blackListFileName ../ref/dm6-blacklist.v2.bed \
&>"$1"_sieve_alignment.log1 && samtools index ./"$1"_final.bam"}' ../rawdata/ATAC_samplename.txtpbs脚本
#!/bin/bash
#PBS -q large_q
#PBS -l nodes=1:ppn=6
#PBS -l mem=32gb
cd /data1/home/Chennan01/RNA-Seq/starrseq/cleanbams
/usr/local/bin/singularity exec ../sifs/deeptools_20231220.sif alignmentSieve --numberOfProcessors 6 --bam ../rawbams/A_control_rep1_sorted.bam --outFile ./A_control_rep1_final.bam --filterMetrics A_control_rep1_sieve_alignment.log --ignoreDuplicates --minMappingQuality 25 --samFlagExclude 260 --ATACshift --blackListFileName ../ref/dm6-blacklist.v2.bed &>A_control_rep1_sieve_alignment.log1 && samtools index ./A_control_rep1_final.bam
/usr/local/bin/singularity exec ../sifs/deeptools_20231220.sif alignmentSieve --numberOfProcessors 6 --bam ../rawbams/A_control_rep2_sorted.bam --outFile ./A_control_rep2_final.bam --filterMetrics A_control_rep2_sieve_alignment.log --ignoreDuplicates --minMappingQuality 25 --samFlagExclude 260 --ATACshift --blackListFileName ../ref/dm6-blacklist.v2.bed &>A_control_rep2_sieve_alignment.log1 && samtools index ./A_control_rep2_final.bam
/usr/local/bin/singularity exec ../sifs/deeptools_20231220.sif alignmentSieve --numberOfProcessors 6 --bam ../rawbams/A_control_rep3_sorted.bam --outFile ./A_control_rep3_final.bam --filterMetrics A_control_rep3_sieve_alignment.log --ignoreDuplicates --minMappingQuality 25 --samFlagExclude 260 --ATACshift --blackListFileName ../ref/dm6-blacklist.v2.bed &>A_control_rep3_sieve_alignment.log1 && samtools index ./A_control_rep3_final.bam
/usr/local/bin/singularity exec ../sifs/deeptools_20231220.sif alignmentSieve --numberOfProcessors 6 --bam ../rawbams/A_HKSM_rep1_sorted.bam --outFile ./A_HKSM_rep1_final.bam --filterMetrics A_HKSM_rep1_sieve_alignment.log --ignoreDuplicates --minMappingQuality 25 --samFlagExclude 260 --ATACshift --blackListFileName ../ref/dm6-blacklist.v2.bed &>A_HKSM_rep1_sieve_alignment.log1 && samtools index ./A_HKSM_rep1_final.bam
/usr/local/bin/singularity exec ../sifs/deeptools_20231220.sif alignmentSieve --numberOfProcessors 6 --bam ../rawbams/A_HKSM_rep2_sorted.bam --outFile ./A_HKSM_rep2_final.bam --filterMetrics A_HKSM_rep2_sieve_alignment.log --ignoreDuplicates --minMappingQuality 25 --samFlagExclude 260 --ATACshift --blackListFileName ../ref/dm6-blacklist.v2.bed &>A_HKSM_rep2_sieve_alignment.log1 && samtools index ./A_HKSM_rep2_final.bam
/usr/local/bin/singularity exec ../sifs/deeptools_20231220.sif alignmentSieve --numberOfProcessors 6 --bam ../rawbams/A_HKSM_rep3_sorted.bam --outFile ./A_HKSM_rep3_final.bam --filterMetrics A_HKSM_rep3_sieve_alignment.log --ignoreDuplicates --minMappingQuality 25 --samFlagExclude 260 --ATACshift --blackListFileName ../ref/dm6-blacklist.v2.bed &>A_HKSM_rep3_sieve_alignment.log1 && samtools index ./A_HKSM_rep3_final.bam结果
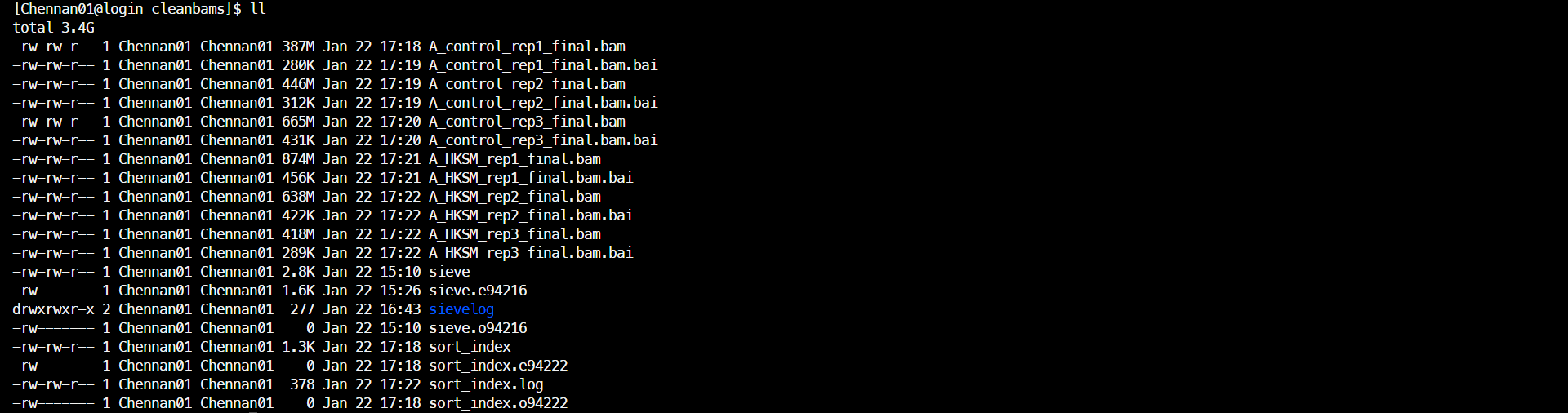
(2)排序并建立索引
pbs脚本
cd /data1/home/Chennan01/RNA-Seq/starrseq/cleanbams
# 排序时屏幕会输出乱码,所以不保存。换行符\n在双引号内,\n后不加空格。换行写要加续行符\
awk '{print "samtools sort \
-@ 2 \
"$1"_final.bam \
-o "$1"_tmp.bam \
2>>sort_index.log \nmv "$1"_tmp.bam "$1"_final.bam \n\
samtools index "$1"_final.bam &>>sort_index.log\n"}' ../rawdata/ATAC_samplename.txt
#!/bin/bash
#PBS -q big_q
#PBS -l nodes=1:ppn=2
#PBS -l mem=16gb
cd /data1/home/Chennan01/RNA-Seq/starrseq/cleanbams
samtools sort -@ 2 A_control_rep1_final.bam -o A_control_rep1_tmp.bam 2>>sort_index.log
mv A_control_rep1_tmp.bam A_control_rep1_final.bam
samtools index A_control_rep1_final.bam &>>sort_index.log
samtools sort -@ 2 A_control_rep2_final.bam -o A_control_rep2_tmp.bam 2>>sort_index.log
mv A_control_rep2_tmp.bam A_control_rep2_final.bam
samtools index A_control_rep2_final.bam &>>sort_index.log
samtools sort -@ 2 A_control_rep3_final.bam -o A_control_rep3_tmp.bam 2>>sort_index.log
mv A_control_rep3_tmp.bam A_control_rep3_final.bam
samtools index A_control_rep3_final.bam &>>sort_index.log
samtools sort -@ 2 A_HKSM_rep1_final.bam -o A_HKSM_rep1_tmp.bam 2>>sort_index.log
mv A_HKSM_rep1_tmp.bam A_HKSM_rep1_final.bam
samtools index A_HKSM_rep1_final.bam &>>sort_index.log
samtools sort -@ 2 A_HKSM_rep2_final.bam -o A_HKSM_rep2_tmp.bam 2>>sort_index.log
mv A_HKSM_rep2_tmp.bam A_HKSM_rep2_final.bam
samtools index A_HKSM_rep2_final.bam &>>sort_index.log
samtools sort -@ 2 A_HKSM_rep3_final.bam -o A_HKSM_rep3_tmp.bam 2>>sort_index.log
mv A_HKSM_rep3_tmp.bam A_HKSM_rep3_final.bam
samtools index A_HKSM_rep3_final.bam &>>sort_index.log结果
(3)过滤报告
每个步骤中过滤掉了多少 reads 可以使用 deeptools 中的 estimateReadFiltering 进行估计。 不需要加 --ATACshift
pbs脚本
#!/bin/bash
#PBS -q large_q
#PBS -l nodes=1:ppn=8
#PBS -l mem=40gb
cd /data1/home/Chennan01/RNA-Seq/starrseq/rawbams
/usr/local/bin/singularity exec ../sifs/deeptools_20231220.sif estimateReadFiltering \
--numberOfProcessors 8 \
--bam A_control_rep1_sorted.bam A_control_rep2_sorted.bam A_control_rep3_sorted.bam A_HKSM_rep1_sorted.bam A_HKSM_rep2_sorted.bam A_HKSM_rep3_sorted.bam \
--sampleLabels A_control_rep1 A_control_rep2 A_control_rep3 A_HKSM_rep1 A_HKSM_rep2 A_HKSM_rep3 \
--outFile ../cleanbams/estimate_filter_report.tsv \
--ignoreDuplicates \
--minMappingQuality 25 \
--samFlagExclude 260 \
--blackListFileName ../ref/dm6-blacklist.v2.bed \
&>../cleanbams/estimate_filter.log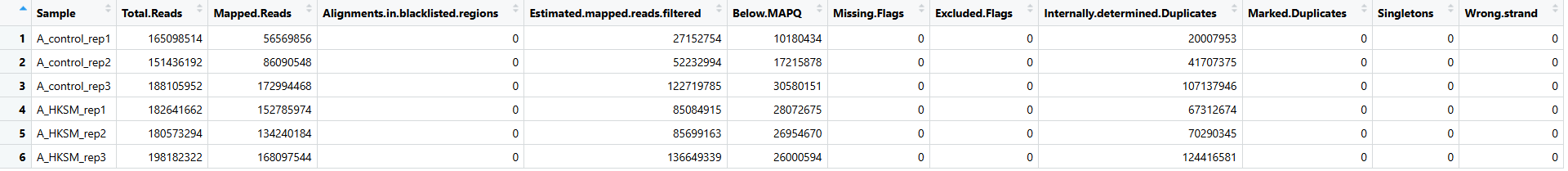
结果
5.call peak(MACS3)
Call Peak用于识别比对的reads富集到基因组的区域
计算有效基因组大小=总长度-N碱基数。(使用Kents tools的faCount)
[Chennan01@login scripts]$ ./faCount -summary ../../../starrseq/ref/dmel-all-chromosome-r6.66.fasta
#seq len A C G T N cpg
total 143726002 41351058 29960672 29928813 41332481 1152978 5893673
prcnt 1.0 0.2877 0.2085 0.2082 0.2876 0.0080 0.0410
143726002-1152978=142573024(1)call peak
-
ATAC-seq:没有 input,不需要 -c
-
--nomodel:改为手动指定偏移和扩展方式。默认会尝试估算样本的片段长度分布(d值)来建模并移动reads,这个模型对于ChIP-seq的转录因子数据很有效。但ATAC-seq的片段长度分布非常特殊,包含核小体游离区(短片段)和核小体缠绕区(长片段)的混合信号,自动模型可能不准。
-
--shift -100:Tn5转座酶在切割DNA时,会在两条链上各留下一个9bp的缺口。当测序读长从切割位点开始,为了还原真实的切割中心,需要将reads的5‘端往回(5’方向)移动。移动的距离并非简单的9bp,而是根据统计学观察,Tn5的切割位点距离其结合的开放区域中心平均约100bp。
-
--extsize 200:在进行了位置校正后,需要将每个read(现在代表一个切割信号)扩展成一个固定长度的片段来模拟开放的染色质区域。ATAC-seq中核小体游离区域(NFR)的典型大小在200bp左右。因此,将校正后的read向两侧扩展至200bp,可以更好地覆盖整个开放区域,从而在堆积计数时形成连续、明显的峰。
-
常见的组合如
-100/200或-75/150,是对“切割位点至开放区域中心距离”和“开放区域典型大小”的估算,可根据实际数据或文献微调。
-
-
--format BAM:即使是 PE 测序,也不能设置 BAMPE,否则会使用 PE 的 fragment size,手动设置失效。-f BAMPE和 --nomodel --shift --extsize是两种互斥的分析策略。前者基于实际片段,后者基于人为校正和定义的虚拟片段。
macs3 callpeak示例
macs3 callpeak -g 2913022398 \ # 有效基因组大小。fasta基因组文件如有叶绿体线粒体序列,需要去掉序列后再-N
-t clean_bams/Diabetic_hs_rep1_final.bam \
--name raw_peaks/Diabetic_hs_rep1 \ # 输出文件的前缀名
--format BAM \
--keep-dup all \ # 由于之前已经去除了 dup,这里选 all
--qvalue 0.05 \
--cutoff-analysis \ # 当启用此选项时,MACS3会分析不同p值阈值下可识别峰的数量或总长度,并输出汇总表格帮助用户选择更合适的阈值。该表格将保存为NAME_cutoff_analysis.txt文件。
--broad \ # 只研究转录因子,用narrow,默认narrow,不用设。还研究其他蛋白broad
--nomodel --shift -100 --extsize 200 \
&>logs/Diabetic_hs_rep1_callpeak_with_macs3.logpbs脚本
cd /data1/home/Chennan01/RNA-Seq/starrseq/rawpeaks
awk '{print "/usr/local/bin/singularity exec ../sifs/macs3_idr_20231218.sif macs3 callpeak -g 142573024 \
-t ../cleanbams/"$1"_final.bam \
--name "$1" \
--format BAM \
--keep-dup all \
--qvalue 0.05 \
--cutoff-analysis \
--nomodel --shift -100 --extsize 200 \
&>"$1"_callpeak.log"}' ../rawdata/ATAC_samplename.txt
#!/bin/bash
#PBS -q large_q
#PBS -l nodes=1:ppn=6
#PBS -l mem=16gb
cd /data1/home/Chennan01/RNA-Seq/starrseq/rawpeaks
/usr/local/bin/singularity exec ../sifs/macs3_idr_20231218.sif macs3 callpeak -g 142573024 -t ../cleanbams/A_control_rep1_final.bam --name A_control_rep1 --format BAM --keep-dup all --qvalue 0.05 --cutoff-analysis --nomodel --shift -100 --extsize 200 &>A_control_rep1_callpeak.log
/usr/local/bin/singularity exec ../sifs/macs3_idr_20231218.sif macs3 callpeak -g 142573024 -t ../cleanbams/A_control_rep2_final.bam --name A_control_rep2 --format BAM --keep-dup all --qvalue 0.05 --cutoff-analysis --nomodel --shift -100 --extsize 200 &>A_control_rep2_callpeak.log
/usr/local/bin/singularity exec ../sifs/macs3_idr_20231218.sif macs3 callpeak -g 142573024 -t ../cleanbams/A_control_rep3_final.bam --name A_control_rep3 --format BAM --keep-dup all --qvalue 0.05 --cutoff-analysis --nomodel --shift -100 --extsize 200 &>A_control_rep3_callpeak.log
/usr/local/bin/singularity exec ../sifs/macs3_idr_20231218.sif macs3 callpeak -g 142573024 -t ../cleanbams/A_HKSM_rep1_final.bam --name A_HKSM_rep1 --format BAM --keep-dup all --qvalue 0.05 --cutoff-analysis --nomodel --shift -100 --extsize 200 &>A_HKSM_rep1_callpeak.log
/usr/local/bin/singularity exec ../sifs/macs3_idr_20231218.sif macs3 callpeak -g 142573024 -t ../cleanbams/A_HKSM_rep2_final.bam --name A_HKSM_rep2 --format BAM --keep-dup all --qvalue 0.05 --cutoff-analysis --nomodel --shift -100 --extsize 200 &>A_HKSM_rep2_callpeak.log
/usr/local/bin/singularity exec ../sifs/macs3_idr_20231218.sif macs3 callpeak -g 142573024 -t ../cleanbams/A_HKSM_rep3_final.bam --name A_HKSM_rep3 --format BAM --keep-dup all --qvalue 0.05 --cutoff-analysis --nomodel --shift -100 --extsize 200 &>A_HKSM_rep3_callpeak.log# 一步获取至少2个样本共享的共识峰
bedtools multiinter -i A_control_rep1.bed A_control_rep2.bed A_control_rep3.bed | \
awk '$4 >= 2' | \
cut -f1-3 > consensus_peaks.bed

 浙公网安备 33010602011771号
浙公网安备 33010602011771号