

转录组分析(五):测序数据批量下载(RNA-Seq)
准备数据
(一)测序数据(.fastq):测序公司数据或从数据库下载
(二)数据信息表(sample.txt):每一个样本名称、所属分组、存储路径
(三)参考基因组序列(genome.fasta)、基因注释(genes.gtf)、蛋白序列(proteins.fasta)
一、下载测序数据(RNA-Seq)
SRA数据库:用于存储高通量测序数据(二代/三代)的数据库
1. 生成下载列表(SRR号)
(1)选择列表:send to→Run Selector→Go
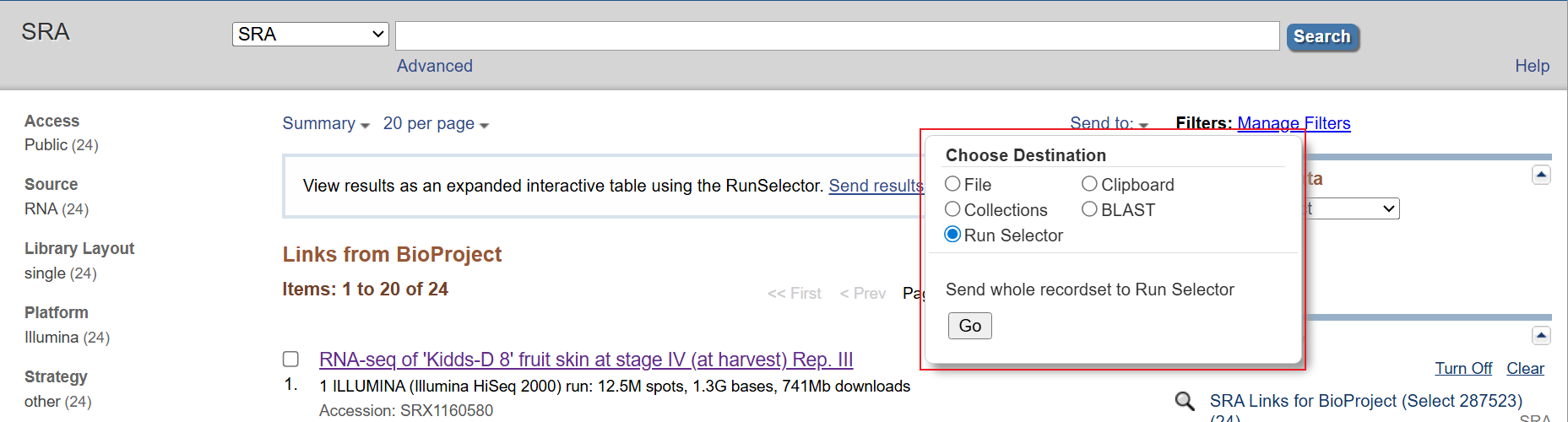
(2)→Accession List
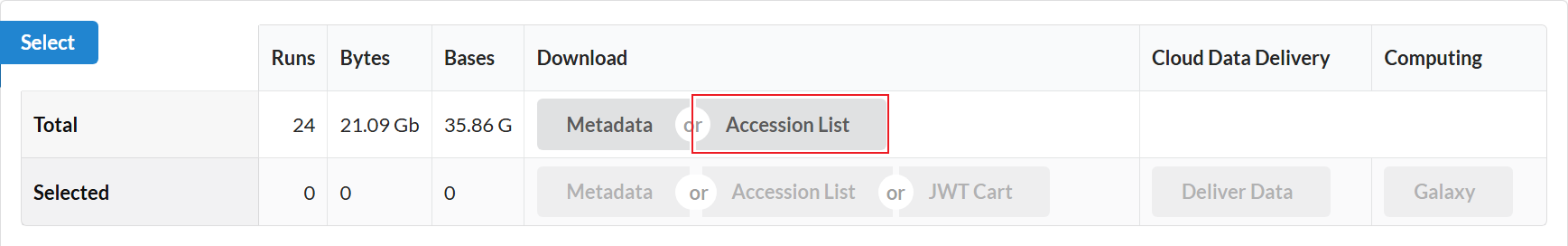
(3)→得到SRR_Acc_List.txt
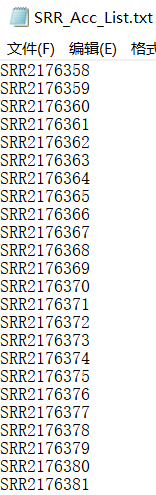
2. 批量下载
# 1.安装下载工具。从https://ftp-trace.ncbi.nih.gov/sra/sdk/中选择历史版本
wget https://ftp-trace.ncbi.nih.gov/sra/sdk/3.0.0/
tar -xvf sratoolkit.3.0.0-centos_linux64.tar.gz
cd sratoolkit.3.0.0-centos_linux64/bin
./vdb-config --interactive //配置
# 2.下载SRA文件(prefetch)、转换(fastq-dump)
prefetch SRR2176381 //单个下载
fastq-dump --split-3 SRR2176381.sra //将SRA文件转换为fastq
awk '{print "prefetch "$1 " &"}' SRR_Acc_List.txt >download.sh //批量下载。加&放后台,并行下载
sh download.sh
#编写脚本批量转换
#!/bin/sh //指定使用 /bin/sh 作为脚本解释器
for i in *sra //循环:遍历当前目录下所有以 ".sra" 结尾的文件
do //循环体开始
echo $i //打印当前正在处理的 SRA 文件名(便于查看进度)
fastq-dump --split-files $i //使用 fastq-dump 工具转换 SRA 文件
done //循环结束
# 3.或者直接下载并转换
awk '{print "fastq-dump --split-3 --gzip "$1" -O /data1/home/Chennan01/RNA-Seq/data"}' SRR_Acc_List.txt >dl.sh
//fastq-dump是NCBI SRA工具包中的命令,用于下载或提取测序数据。
//--split-3:将配对端测序数据分割为两个独立的fastq文件(对应左右端读取)。
//--gzip:输出 gz 格式的压缩文件,以节省空间。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号