

bam文件softclip , hardclip ,markduplicate的探究
测序产生的bam文件,有一些reads在cigar值里显示存在softclip,有一些存在hardclip,究竟softclip和hardclip是怎么判断出来的,还有是怎么标记duplicate的reads的,我怀着这些问题进行了探究。
测试步骤
- 编辑两个bed文件,分别含有我们需要的read1和read2位置,这里每个文件包含两条read1或者两条read2,read1、read2一对作为原始的reads(序列名primer_pri),另一对作为截取的材料(这里取序列名为other)
- 使用bedtools getfasta,从参考基因组获得reads的序列,将read2反向互补。将原始reads放入两个文件,一个test_R1.fa,一个test_R2.fa
- 在test_R1.fa中添加其它修改过的原始reads,并在test_R2.fa中也添加相应的read2,不过read2不修改
read1名称如下

- primer_pri:原始read
- pimer_duplicate1:primer_pri的重复,一模一样
- pimer_duplicate2:read1 primer_pri去掉5‘两个碱基
- pimer_duplicate3:read1 primer_pri去掉5’两个碱基,再去掉3'两个碱基
- pimer_changeR2Termi5base:read1修改了read2 5‘端的碱基
- primer_halfother:read1截掉后面reads,用other的5‘部分reads补全
- pimer_change3Termi5base_change5Termi5base_sametwo:read1和read2一样,并且5'端和3‘端都改变了5个碱基
- pimer_change3Termi5base_change5Termi5base:read1 5'端和3‘端都改变了5个碱基,但是read2保留primer_pri的read2
结果
softclip和hardclip
其中
- primer_halfother read1 82M65S,有SA tag,SA:Z:chr12,5378700,+,79S68M,60,0
- pimer_change3Termi5base_change5Termi5base_sametwo 两条reads均为5S137M5S
- pimer_change3Termi5base_change5Termi5base read1 5S137M5S
- primer_halfother read1 79H68M ,有SA tag,SA:Z:chr12,5378502,+,82M65S,60,0
结论(部分分析参考SAMv1.pdf文件)
- 对于map到一个位置的read,两端map不上的叫做clip ,map到一个位置的情况下以softclip显示(比如 pimer_change3Termi5base_change5Termi5base_sametwo和 pimer_change3Termi5base_change5Termi5base read1)
- 对于嵌合比对的read(可以map到多个区域,并且这比对上的区域很大部分非overlap),比如primer_halfother read1比对上两个位置,一个比对到chr12 : 5378502,一个比对到chr12:5378700,并且两次hit的位置的碱基overlap少,产生的这种情况是因为read前一部分比对到了前者,而后一部分又可以比对到了后者,因此无论比对到任何一个位置都这条read都是部分match(这种叫做Chimeric alignment/嵌合比对)。
- 嵌合比对的read,有一条是最优的read,因为我们map的时候设置了-M参数,因此认为较短的split的reads断定为优,这里是的62 clip 的hit断定为优。因此65个比对不上的显示为softclip,而另外一个hit,79 clip显示为hard clip,序列中不显示,并且存入0x800(supplementary alignment flag)
- 为什么82M65S对应的是79H68M呢,理论上应该是82H65M才对,这是因为这里两个比对有三个碱基的overlap,所以前面有65+3个match,后面有79+3个match(制造reads的时候碰巧截取的primer read 3'端三个碱基和截取的other read 5‘部分read 三个碱基一样)
- 这种嵌合比对的reads含有SA tag
duplicate
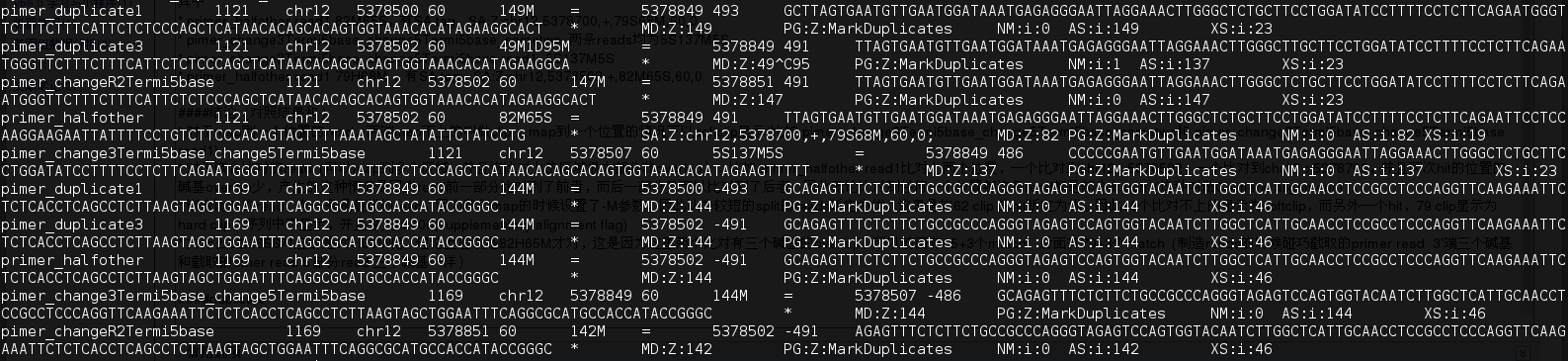
其中mark为duplicate 的reads 对(duplicate 是按fragment算) 有 primer duplicate1,primer_duplicate3,pimer_changeR2Termi5base,primer_halfother(82M65S,144M(未改的read2)),pimer_change3Termi5base_change5Termi5base
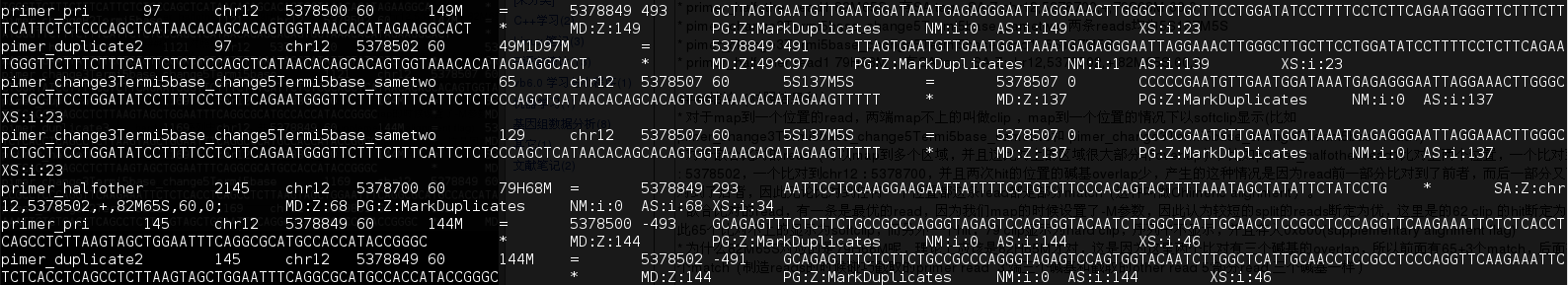
不属于duplicate的有
primer_pri,pimer_duplicate2,primer_halfother(79H68M,一条),pimer_change3Termi5basechange5Termi5base_sametwo
结论
- fragment的start和end一样(read1和read2(因为read2是测对链的,reads的5‘端都是fragment的末端)的5’的位置都相同)判断为duplicate,只取最优的不标记为duplicate
- primer_pri的duplicate是 primer duplicate1, primer_halfother
- pimer_duplicate2的duplicate是primer_duplicate3,pimer_changeR2Termi5base,pimer_change3Termi5base_change5Termi5base
- 没有duplicate的是primer_halfother(79H68M,一条),pimer_change3Termi5basechange5Termi5base_sametwo
- pimer_change3Termi5basechange5Termi5base_sametwo 5'有5 softclip,map的位置从M的碱基算起(见图),所以它没有duplicate

 浙公网安备 33010602011771号
浙公网安备 33010602011771号