- 如果是都在编码区,那么肯定是严格的翻译关系。
- 对于DNA我们需要确定阅读框在哪,可能是0,1,2。
- DNA是有反链的,也有可能是反链的0,1,2,也有可能对应上。
- 最后比较重合部分,也就是最长公共子串。
![image]()
def get_reverse_complement(dna_sequence):
"""
获取DNA序列的反向互补链
参数:
dna_sequence: DNA序列字符串
返回:
反向互补链序列字符串
"""
complement_map = {'A': 'T', 'T': 'A', 'C': 'G', 'G': 'C'}
reverse_seq = dna_sequence[::-1] # 反向
reverse_complement = ''.join(complement_map.get(base, base) for base in reverse_seq.upper())
return reverse_complement
def translate_dna(dna_sequence, start_index=0):
"""
从指定起始位置翻译DNA序列为蛋白质序列
参数:
dna_sequence: DNA序列字符串
start_index: 起始位置(0, 1, 2)
返回:
蛋白质序列字符串
"""
# 密码子表 (标准遗传密码)
codon_table = {
'ATA': 'I', 'ATC': 'I', 'ATT': 'I', 'ATG': 'M',
'ACA': 'T', 'ACC': 'T', 'ACG': 'T', 'ACT': 'T',
'AAC': 'N', 'AAT': 'N', 'AAA': 'K', 'AAG': 'K',
'AGC': 'S', 'AGT': 'S', 'AGA': 'R', 'AGG': 'R',
'CTA': 'L', 'CTC': 'L', 'CTG': 'L', 'CTT': 'L',
'CCA': 'P', 'CCC': 'P', 'CCG': 'P', 'CCT': 'P',
'CAC': 'H', 'CAT': 'H', 'CAA': 'Q', 'CAG': 'Q',
'CGA': 'R', 'CGC': 'R', 'CGG': 'R', 'CGT': 'R',
'GTA': 'V', 'GTC': 'V', 'GTG': 'V', 'GTT': 'V',
'GCA': 'A', 'GCC': 'A', 'GCG': 'A', 'GCT': 'A',
'GAC': 'D', 'GAT': 'D', 'GAA': 'E', 'GAG': 'E',
'GGA': 'G', 'GGC': 'G', 'GGG': 'G', 'GGT': 'G',
'TCA': 'S', 'TCC': 'S', 'TCG': 'S', 'TCT': 'S',
'TTC': 'F', 'TTT': 'F', 'TTA': 'L', 'TTG': 'L',
'TAC': 'Y', 'TAT': 'Y', 'TAA': '*', 'TAG': '*',
'TGC': 'C', 'TGT': 'C', 'TGA': '*', 'TGG': 'W',
}
protein = ""
dna_upper = dna_sequence.upper()
# 从指定起始位置开始,每次取3个碱基
for i in range(start_index, len(dna_upper) - 2, 3):
codon = dna_upper[i:i+3]
if len(codon) == 3:
amino_acid = codon_table.get(codon, '?')
protein += amino_acid
return protein
def find_overlap(seq1, seq2):
"""
查找两个序列的最长公共子串
参数:
seq1: 序列1
seq2: 序列2
返回:
最长公共子串,起始位置(seq1中),起始位置(seq2中),长度
"""
m = len(seq1)
n = len(seq2)
max_len = 0
end_pos_seq1 = 0
end_pos_seq2 = 0
# 创建动态规划表
dp = [[0] * (n + 1) for _ in range(m + 1)]
for i in range(1, m + 1):
for j in range(1, n + 1):
if seq1[i-1] == seq2[j-1]:
dp[i][j] = dp[i-1][j-1] + 1
if dp[i][j] > max_len:
max_len = dp[i][j]
end_pos_seq1 = i
end_pos_seq2 = j
if max_len == 0:
return "", 0, 0, 0
# 提取最长公共子串
lcs = seq1[end_pos_seq1 - max_len:end_pos_seq1]
start_seq1 = end_pos_seq1 - max_len
start_seq2 = end_pos_seq2 - max_len
return lcs, start_seq1, start_seq2, max_len
def find_all_matches(seq1, seq2, min_length=3):
"""
查找两个序列之间所有长度≥min_length的匹配子串
参数:
seq1: 序列1
seq2: 序列2
min_length: 最小匹配长度
返回:
匹配子串列表,每个元素为字典,包含起始位置、长度和序列
"""
matches = []
# 使用滑动窗口方法查找所有匹配
for i in range(len(seq1)):
for j in range(len(seq2)):
k = 0
while (i + k < len(seq1) and
j + k < len(seq2) and
seq1[i+k] == seq2[j+k]):
k += 1
if k >= min_length:
matches.append({
'seq1_start': i,
'seq2_start': j,
'length': k,
'sequence': seq1[i:i+k]
})
# 如果没有找到匹配,返回空列表
if not matches:
return []
# 去重,保留最长的匹配
matches.sort(key=lambda x: x['length'], reverse=True)
unique_matches = []
seen_positions = set()
for match in matches:
pos_key = (match['seq1_start'], match['seq2_start'])
if pos_key not in seen_positions:
seen_positions.add(pos_key)
unique_matches.append(match)
return unique_matches
def compare_all_frames(dna_sequence, protein_sequence):
"""
主函数:翻译6个阅读框并与给定蛋白质序列比较
参数:
dna_sequence: DNA序列
protein_sequence: 蛋白质序列
返回:
包含比较结果的字典
"""
results = {}
# 获取反向互补链
reverse_complement = get_reverse_complement(dna_sequence)
# 定义链名称
strands = [
("正向链", dna_sequence),
("反向互补链", reverse_complement)
]
# 对每条链的3个阅读框进行翻译
for strand_name, strand_seq in strands:
for frame in range(3):
key = f"{strand_name}_阅读框{frame}"
translated_protein = translate_dna(strand_seq, frame)
# 计算与目标蛋白质序列的最长公共子串
longest_overlap, start_trans, start_target, length = find_overlap(translated_protein, protein_sequence)
# 计算相似度
similarity = 0
if len(translated_protein) > 0 and len(protein_sequence) > 0:
min_len = min(len(translated_protein), len(protein_sequence))
similarity = (length / min_len) * 100 if min_len > 0 else 0
# 查找所有重合部分
all_matches = find_all_matches(translated_protein, protein_sequence, min_length=3)
# 存储结果
results[key] = {
'translated_protein': translated_protein,
'longest_overlap': longest_overlap,
'overlap_length': length,
'similarity_percent': similarity,
'all_matches': all_matches,
'frame': frame,
'strand': strand_name,
'translation_start': start_trans if longest_overlap else None,
'target_start': start_target if longest_overlap else None
}
return results
def print_detailed_results(dna_seq, protein_seq, results):
"""
打印详细的比较结果
参数:
dna_seq: DNA序列
protein_seq: 蛋白质序列
results: 比较结果字典
"""
print("=" * 80)
print("DNA序列分析结果 (6个阅读框)")
print("=" * 80)
print(f"DNA序列长度: {len(dna_seq)}")
print(f"DNA序列: {dna_seq}")
print(f"DNA反向互补序列: {get_reverse_complement(dna_seq)}")
print(f"目标蛋白质序列长度: {len(protein_seq)}")
print(f"目标蛋白质序列: {protein_seq}")
print()
# 按链分组显示结果
strands = ["正向链", "反向互补链"]
for strand in strands:
print(f"{strand}:")
print("-" * 40)
for frame in range(3):
key = f"{strand}_阅读框{frame}"
data = results[key]
print(f" 阅读框 {frame}:")
print(f" 翻译的蛋白质序列: {data['translated_protein']}")
print(f" 翻译序列长度: {len(data['translated_protein'])}")
if data['longest_overlap']:
print(f" 最长重合部分: {data['longest_overlap']}")
print(f" 重合长度: {data['overlap_length']}")
print(f" 在翻译序列中起始位置: {data['translation_start']}")
print(f" 在目标序列中起始位置: {data['target_start']}")
print(f" 相似度: {data['similarity_percent']:.2f}%")
else:
print(f" 最长重合部分: 无")
print(f" 重合长度: 0")
print(f" 相似度: 0%")
if data['all_matches']:
print(f" 所有重合部分 (长度≥3):")
for i, match in enumerate(data['all_matches'][:3], 1): # 只显示前3个
print(f" {i}. 位置: 翻译序列[{match['seq1_start']}]-目标序列[{match['seq2_start']}], "
f"长度: {match['length']}, 序列: {match['sequence']}")
if len(data['all_matches']) > 3:
print(f" ... 以及 {len(data['all_matches']) - 3} 个其他匹配")
else:
print(f" 所有重合部分 (长度≥3): 无")
print()
def find_best_match(results, protein_sequence):
"""
在所有阅读框中找到与目标蛋白质序列最佳匹配的结果
参数:
results: 比较结果字典
protein_sequence: 目标蛋白质序列
返回:
最佳匹配的结果
"""
best_match = None
best_length = 0
for key, data in results.items():
if data['overlap_length'] > best_length:
best_length = data['overlap_length']
best_match = (key, data)
return best_match
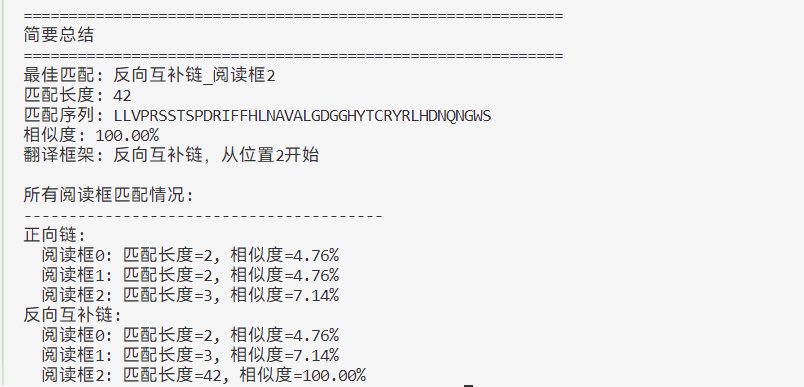
def print_summary(dna_seq, protein_seq, results):
"""
打印简要总结,突出显示最佳匹配
参数:
dna_seq: DNA序列
protein_seq: 蛋白质序列
results: 比较结果字典
"""
print("=" * 60)
print("简要总结")
print("=" * 60)
# 找到最佳匹配
best_match_info = find_best_match(results, protein_seq)
if best_match_info:
best_key, best_data = best_match_info
print(f"最佳匹配: {best_key}")
print(f"匹配长度: {best_data['overlap_length']}")
print(f"匹配序列: {best_data['longest_overlap']}")
print(f"相似度: {best_data['similarity_percent']:.2f}%")
# 显示翻译框架的详细信息
if best_data['strand'] == "正向链":
print(f"翻译框架: 从DNA位置{best_data['frame']}开始")
else:
print(f"翻译框架: 反向互补链,从位置{best_data['frame']}开始")
else:
print("未找到匹配")
print()
# 显示所有阅读框的匹配长度
print("所有阅读框匹配情况:")
print("-" * 40)
strands = ["正向链", "反向互补链"]
for strand in strands:
print(f"{strand}:")
for frame in range(3):
key = f"{strand}_阅读框{frame}"
data = results[key]
print(f" 阅读框{frame}: 匹配长度={data['overlap_length']}, 相似度={data['similarity_percent']:.2f}%")
# 示例使用
if __name__ == "__main__":
# 示例1: 短序列
print("示例1: 短序列分析")
print("=" * 60)
#|
# 示例DNA序列
dna_example = "GGACCAGCCGTTTTGGTTGTCATGCAGCCGGTAGCGGCAGGTGTAGTGACCTCCATCCCCCAGAGCCACCGCGTTCAGGTGAAAGAAGATGCGATCTGGGCTGGTGCTGCTCCTGGGTACCAGCAGCT"
# 示例蛋白质序列
protein_example = "PPPPVLMHHGESSQVLHPGNKVTLTCVAPLSGVDFQLRRGEKELLVPRSSTSPDRIFFHLNAVALGDGGHYTCRYRLHDNQNGWSGDSAPVELILSDETLPAPEFSPEPESGRALRLRCLAPLEGARF"
# 比较所有6个阅读框
comparison_results = compare_all_frames(dna_example, protein_example)
# 打印详细结果
print_detailed_results(dna_example, protein_example, comparison_results)
# 打印简要总结
print_summary(dna_example, protein_example, comparison_results)



 浙公网安备 33010602011771号
浙公网安备 33010602011771号