

寻找U2OS中表达的基因及其promoter并用于后续annotation
方法1.RNA-seq得到不同表达程度基因
方法2. 直接download U2OS_gene.csv https://cancer.sanger.ac.uk/cell_lines/download
最开始excel直接选用25%最高和25%最低,U2OS细胞共~16000基因,故复制前4000行的gene symbol并存为txt;
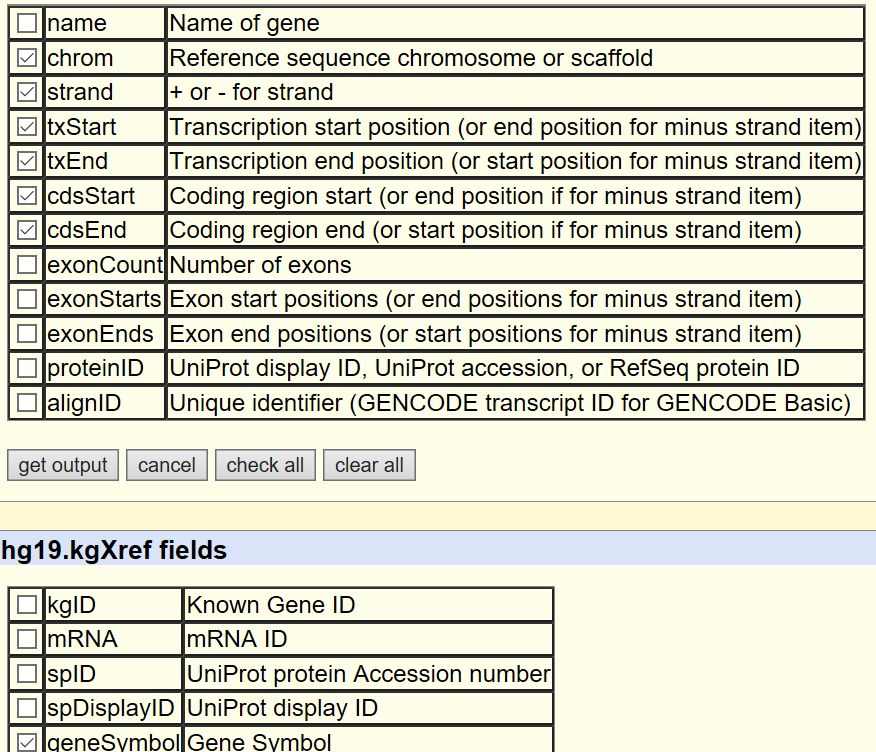
table browser下载'group:Genes and gene prediction; track:UCSC genes; outpu format:selected fileds from primary and related tables' then getoutput,如下图选择
问题出现在grep -wFf 25%_most_highly_expressed_gene_name.txt hg19_geneid_genesymbol.txt > 25%_most_highly_expressed_geneid.txt总是没有输出
trouble shooting首先检查代码,自定义两个文件1.txt 2.txt然后 grep -wFf 1.txt 2.txt成功;
然后检查输入文件hg19_geneid_genesymbol.txt,自定义基因文件(随便选几个U2OS/non-U2OS基因 vi gene.txt),grep -wFf gene.txt hg19_geneid_genesymbol.txt成功;
最后发现问题出在25%_most_highly_expressed_gene_name.txt,最开始得到这个文件是直接从csv中copy and paste,但csv是 comma delimited,所以复制事实上连,一起复制了
#$ head U2OS_genes.csv

#$ head 25%_most_highly_expressed_gene_name.txt

事实上在做grep的时候是“ ,MED6, ”,因此无法匹配 hg19_geneid_genesymbol.txt,这也是为什么grep 'MED6' hg19_geneid_genesymbol.txt 可以work的原因
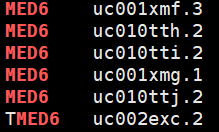
正确做法
#0.6是第4000个基因的zscore
awk -F',' '$5 > 0.6 {print $3}' U2OS_genes.csv > 25%_most_highly_expressed_gene_name.txt
0.6有点过低,做zscore散点图可发现用2更为合理
awk -F',' '$5 > 2 {print $3}' U2OS_genes.csv > highest_expressed_gene_name.txt

更为准确的方法是用R quantile得到合适Z score筛选得到most_expressed 和 least_expressed
grep -wFf highest_expressed_genesym.txt gene_hg19.bed > highest_expressed_gene.bed
PS:head gene_hg19.bed
![]()

一个基因有不同的cds
https://www.jianshu.com/p/cc5cd7053d6e

 浙公网安备 33010602011771号
浙公网安备 33010602011771号