3.测序SRA数据下载与转换
一、官方下载工具Sratoolkit安装
推荐使用conda直接安装,避免配置环境的麻烦,但sratoolkit在conda镜像中的包名为sra-tools
1 conda install -y -c bioconda sra-tools
二、SRA文件下载地址获取
1.NCBI GEO数据库下载地址
1 https://www.ncbi.nlm.nih.gov/geo/
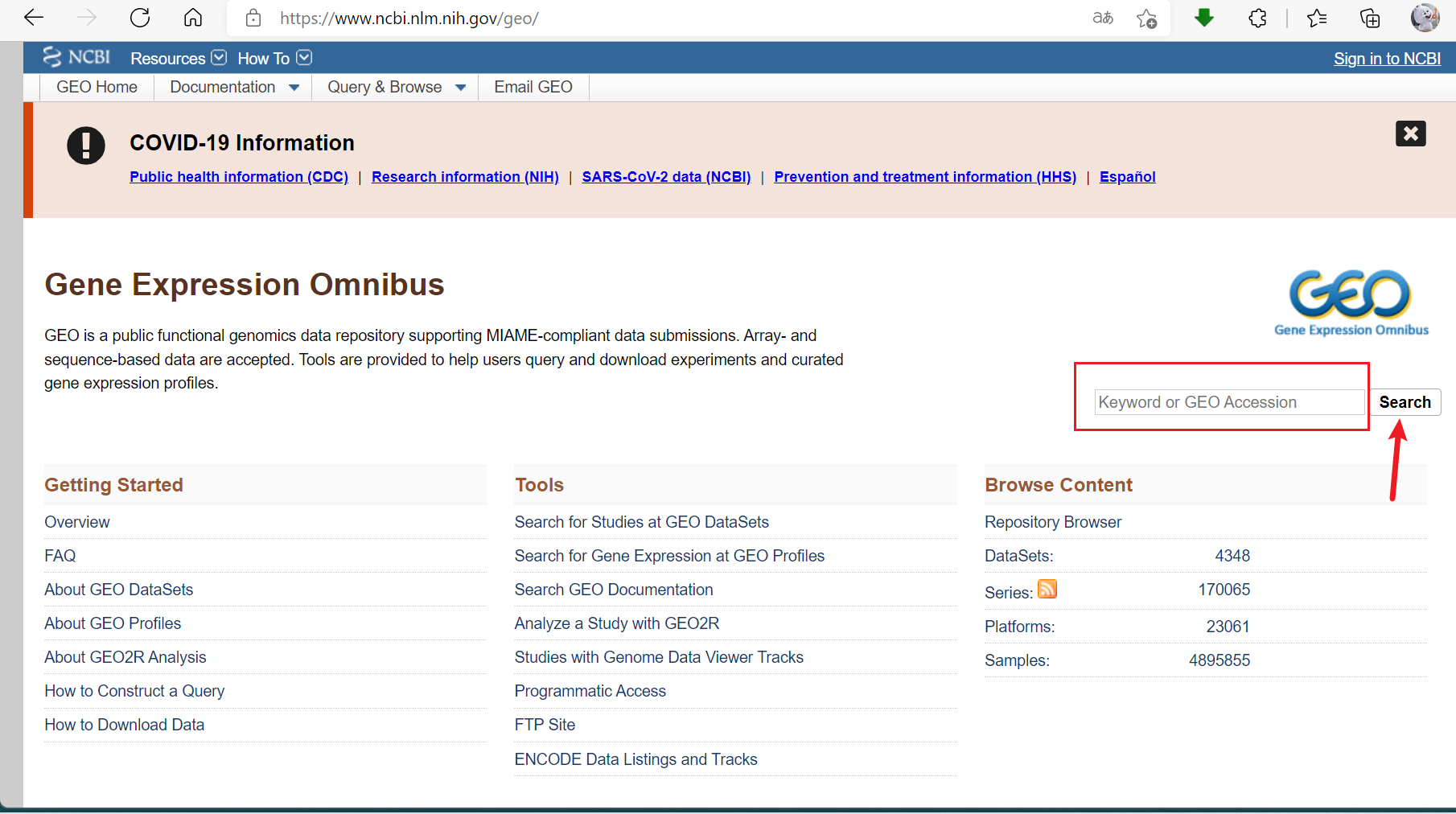
2.输入GEO Accession(如GSE52778),点击搜索,找到测序SRA文件
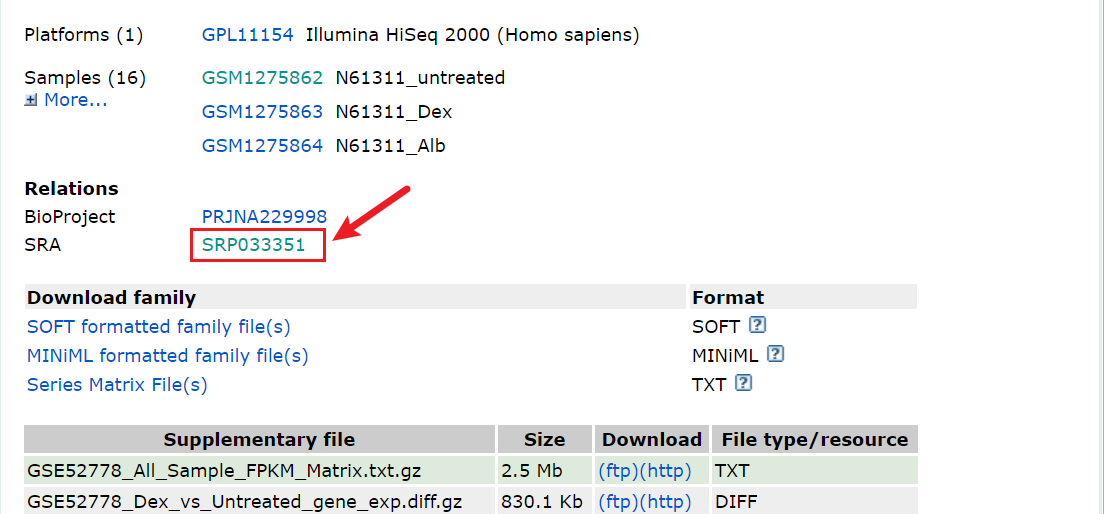
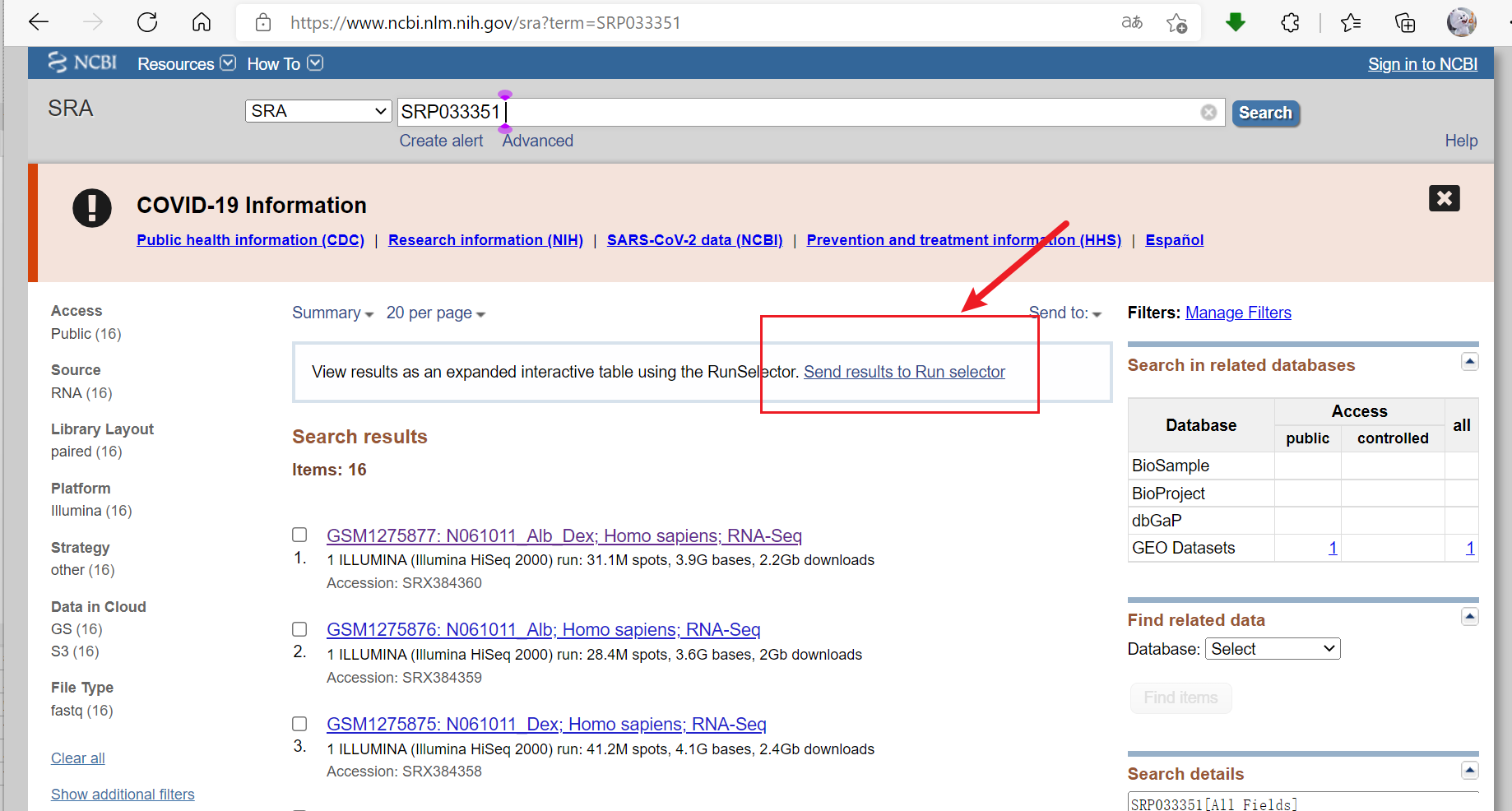
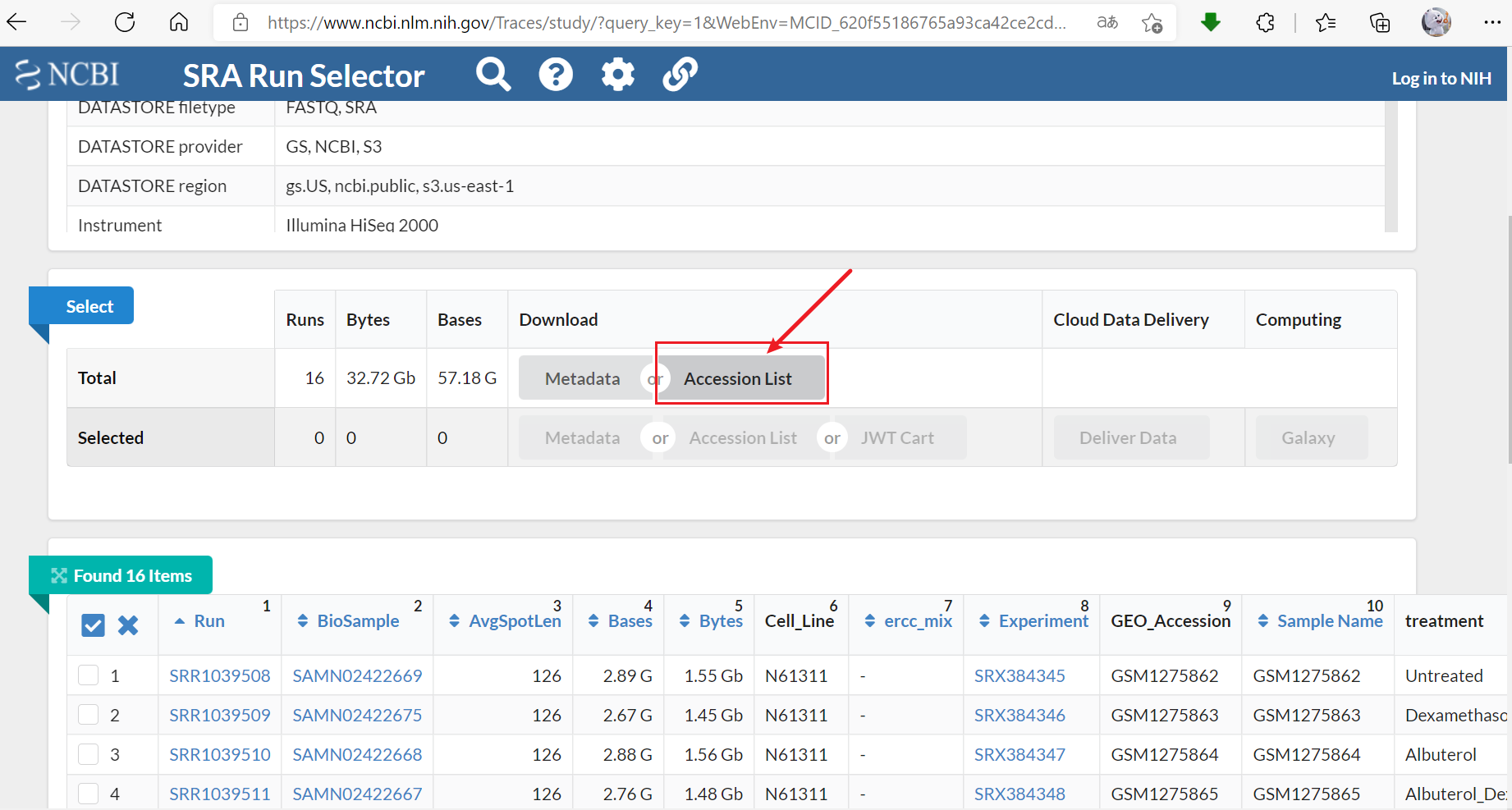
3. 下载Accession List:一个txt文件包含所有的测序样本名称,均对应于上图左下角的Run

三、数据下载
使用sratoolkit提供的prefetch工具下载,官方文档中说明prefetch能直接根据txt文件下载,但不晓得哪边出毛病报错了,自己编写循环下载,脚本如下:
1 #!/bin/bash 2 3 #id.txt即为下载的Accession List 4 5 for i in $(cat id.txt) 6 do 7 echo "正在下载文件".$i 8 prefetch $i 9 done 10 echo "文件下载完毕!!" 11 12 #提取下载的文件夹中的.sra文件并删除源文件夹 13 14 pwd=$(pwd) 15 for i in $(ls) 16 do 17 new_pwd="$pwd/$i" 18 if [ -d $new_pwd ];then 19 mv $new_pwd/* $pwd 20 rm -rf $new_pwd 21 fi 22 done
四、数据转换
使用sratoolkit提供的fastq-dump将下载的SRA数据转换成fastq格式
1 #!/bin/bash 2 for i in SRR* 3 do 4 fastq-dump --gzip --split-3 -O './fastq' $i 5 done
参数说明:
1 --gzip 生成压缩的gz格式fastq文件,以节省磁盘空间 2 -O 输出文件路径 3 --split-files 对read进行拆分,默认不对reads进行拆分, 对于单端测序(SE)没有出现问题.但是对于双端测序(PE)而言,就会把原本的两条reads合并成一个 4 --split-spot: 将双端测序分为两份,但是都放在同一个文件中 5 --split-files: 将双端测序分为两份,放在不同的文件,但是对于一方有而一方没有的reads直接丢弃 6 --split-3 : 将双端测序分为两份,放在不同的文件,但是对于一方有而一方没有的reads会单独放在一个文件夹里

 浙公网安备 33010602011771号
浙公网安备 33010602011771号