


记录单细胞转录组分析入门及注意事项
半个月前接触到一个新项目,因此有了新的分析需求--单细胞转录组分析。首先是熟悉单细胞测序技术的原理及应用,因为此前有过处理组学数据的经验,因此整个学习到分析上手其实是很快的。关于细胞亚群分群注释其实很大程度上是人为决定的,基于表达趋势进行PCA降维分群,筛选标志marker基因进行标注细胞类型。这里面有非常多的坑。经过半个月的从0到1摸索,总算对单细胞转录组分析流程有了一些浅薄理解。以下为使用Seurat包进行单细胞转录组分析的流程及注意事项总结。
参考资料:官网教程https://satijalab.org/seurat/
b站科研鸡官方up的单细胞测序入门三集视频https://www.bilibili.com/video/BV1Dh4y1D7m6?spm_id_from=333.788.videopod.sections&vd_source=79f3f2c3a0e309f88b9eb7edcf162c71
Jimmy老师单细胞亚群注释https://www.bilibili.com/video/BV1mE421w7Te/?spm_id_from=333.788.top_right_bar_window_custom_collection.content.click&vd_source=79f3f2c3a0e309f88b9eb7edcf162c71
Seurat对象结构的理解https://b23.tv/G3UTN0r
一、环境配置
环境安装配置我是采用比较偷懒的方式通过anaconda官网查询需要的r包然后通过conda进行安装,激活环境后直接使用R进行分析。但是这样有一个问题就是我们需要通过看结果文件来调整参数,没有交互式的图形界面就很麻烦,这时候需要R-studio server,由于服务器权限问题,配置不方便,所以我是通过conda安装需要的包到环境中使用,然后远程连接R-studio server随时通过浏览器查看输出结果。
#如果冲突就分开安装,一般没问题
conda create -n seurat conda-forge::r-seurat conda-forge::r-dplyr conda-forge::r-patchwork conda-forge::r-ggplot2 conda-forge::r-harmony -y
#使用及加载所需R包,所需包加载不报错可以进行后续分析
conda activate seurat
R
library(dplyr)
library(Seurat)
library(patchwork)
library(ggplot2)
library(harmony)PS:留了一点点小坑待以后解决,服务器上的R-studio如果能直接配置好环境就不需要这么麻烦。
二、R的基础操作
由于先前处理数据都是通过shell写脚本处理高通量数据,所以很少用到R,这里需要先学习一些R的基础操作,理解R的数据结构,处理方式。不赘述,自行google。
三、认识你的数据类型
1、单细胞数据下机数据类似bulk转录组分析输出的count矩阵,有过bulk分析经验的都知道,样本中的大部分基因其实是不表达,就是表达量为0,从数据存储分析的角度来说,是没有必要记录的,特别是单细胞转录组数据,每一行是基因,而列是细胞,一个单细胞测序样本往往包含上万个细胞,以人类3w基因为例,这个矩阵规模行列都过万了,但是其中大部分数据都是0,这种矩阵就是稀疏矩阵,针对稀疏矩阵可以通过记录行列坐标及对应表达值,来优化存储。所以我们拿到的测序公司给的数据类型,往往就是这样的数据,而不是直接给表达矩阵。它包含三个文件,barcodes.tsv.gz、features.tsv.gz、matrix.mtx.gz,我们需要检查features行在所有样本中是不是统一。这里就发现有个别样本,他的行不统一,这个造成的原因是上游比对参考基因组选取的基因组版本不统一,需要联系公司重新给数据,或者自己去进行所有原始数据的上游分析,选用同一个版本基因组就不会有这个问题。
setwd("/path2yourdata/data")
rm(list = ls())
#############################多样本的稀疏矩阵读取及合并分析##################################
pbmc.data <- Read10X(data.dir = "pbmc.matrix")
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc", min.cells = 3, min.features = 200)
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
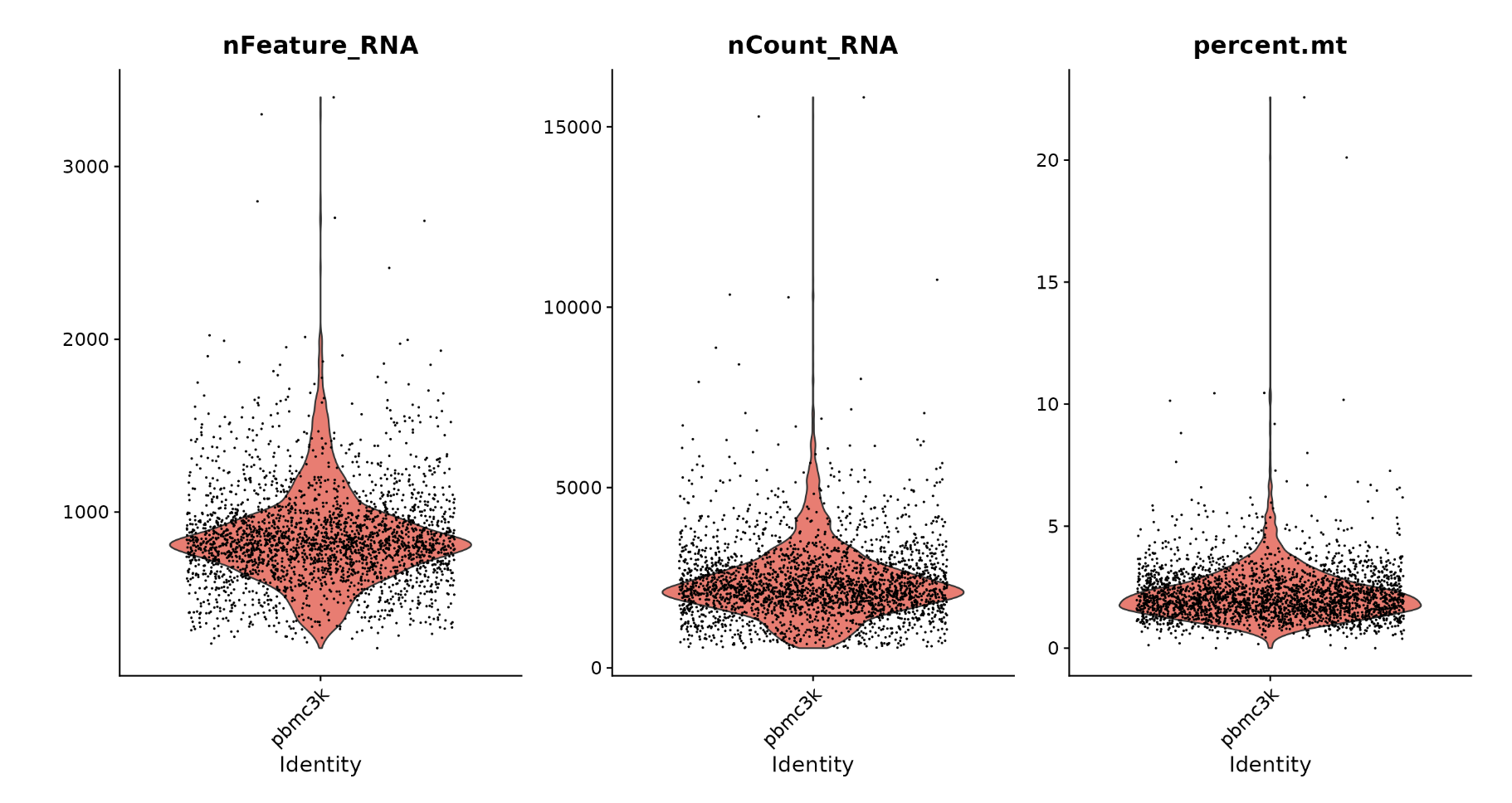
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
pbmc <- subset(pbmc, subset = nFeature_RNA > 500 & nFeature_RNA < 2500 & nCount_RNA < 10000 & percent.mt < 30)
pbmc2.data <- Read10X(data.dir = "pbmc2.matrix")
pbmc2 <- CreateSeuratObject(counts = pbmc2.data, project = "pbmc2", min.cells = 3, min.features = 200)
pbmc2[["percent.mt"]] <- PercentageFeatureSet(pbmc2, pattern = "^MT-")
VlnPlot(pbmc2, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
pbmc2 <- subset(pbmc2, subset = nFeature_RNA > 500 & nFeature_RNA < 2500 & nCount_RNA < 10000 & percent.mt < 30)
scelist = list()
scelist[[1]] = pbmc
scelist[[2]] = pbmc2
s = c('pbmc','pbmc2')
names(scelist) = s
sce.all <- merge((scelist[[1]]), y = scelist[-1], add.cell.ids = s)
#############################多样本的稀疏矩阵读取及合并分析##################################
###合并代码看不懂的可以去看科研鸡的视频里面有讲解,
###另外这个读取方式我感觉也不是很智能,应该可以写个循环读取会更好,留待以后优化吧
###样本少还可以一个一个写,样本多了就麻烦2、除了这种标准格式的单细胞数据,还有给出count矩阵的数据,例如通过GEO数据库下载时,经常会碰到作者只提供了*.count.txt.gz的情况,这种情况直接解压数据,通过R的函数read.table()读入即可。seurat包会将矩阵构造成为seurat对象。需要多理解这个seurat对象数据格式,后续的处理都在这个基础上。
pbmc.data <- read.table("./GSM12345.count.txt")
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc", min.cells = 3, min.features = 200)
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
pbmc <- subset(pbmc, subset = nFeature_RNA > 500 & nFeature_RNA < 5000 & nCount_RNA < 10000 & percent.mt < 50)
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)四、数据质控
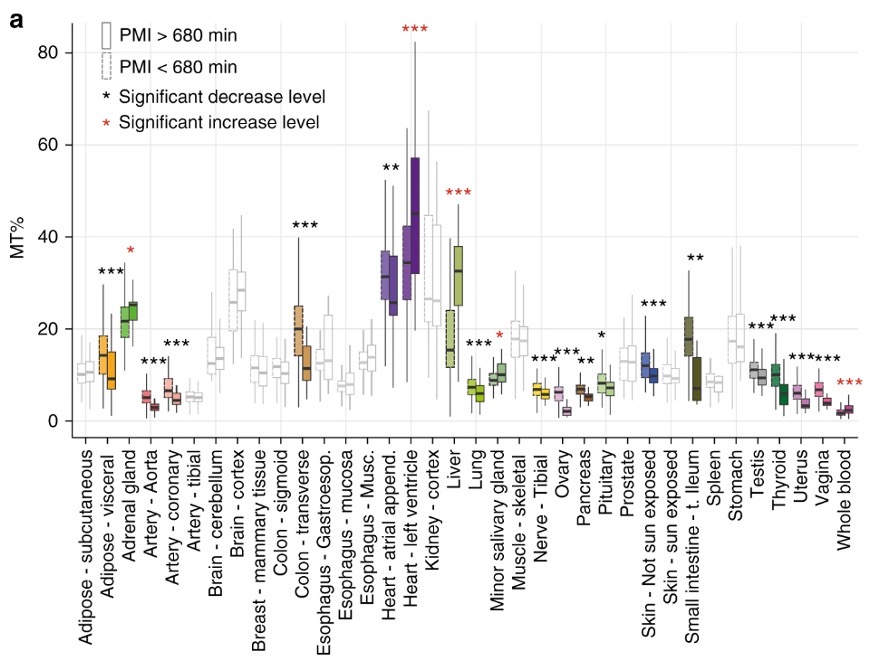
数据质控有几个参数,一般是nFeature_RNA,nCount_RNA,可以按小提琴图掐头去尾,对数据质量要求严格可以将nFeature_RNA范围设置更窄,将峰值包含在中间位置即可,另外线粒体比例需要根据研究的组织类型以及数据的情况进行过滤,不同组织的线粒体本底含量不一样,所以通过线粒体基因筛选死细胞的标准也需要调整(细胞处于濒死状态时,线粒体基因会异常高表达,所以这个表达的比例可以判断细胞的活性状态,死细胞的基因表达模式会和正常组织不一样,会对后续分析造成影响因此需要过滤,另外,在测序时由于测序原理的精度问题,可能会有多胞问题,这里可以通过nFeatures和nCount进行控制,另外还可以引入去多胞的算法通过计算多胞可能性作为参考进行一定比例的多胞过滤)。下面是一篇发表在NC(doi.org/10.1038/s41467-017-02772-x)上的文章,研究了不同人体组织的线粒体基因的本底含量,可以作为一个参考。以下面单一数据集为例,我倾向于更严格的过滤nFeature_RNA控制为500到2000之间,nCount_RNA控制在10000甚至8000以下都可以,mt就在5%以下就好。这里的nFeature的峰值是后面选取高变的一个关键参考。
五、数据降维聚类处理
多个样本的分析涉及到批次效应,需要通过Harmony算法去除批次效应,使原本应该聚为一类的同一细胞类型不因批次效应而聚成不同簇。
PS:留坑,数据降维的过程涉及矩阵特征值分解,等有空时再去了解一些数学原理。
降维分析的过程涉及到3个关键参数,找高变的nfeatures、聚类维度dims以及分辨率,高变基因依据前面的nFeature_RNA的峰值,比如这里我会倾向于使用nfeatures = 1000而不是默认的2000,2000会引入很多的噪音。另外维度的确定可以先看一下ndims = 30,依据肘部图的拐点选取,后续确认选取dims = 1:24,resolution的参数只会影响细胞群是标注成多少个cluster,例如选取0.2-0.8都可以尝试一下,不确定选多少时先用0.5,resolution的参数调整不会影响umap图的形状。
sce.all <- NormalizeData(sce.all, normalization.method = "LogNormalize", scale.factor = 10000)
sce.all <- FindVariableFeatures(sce.all, selection.method = "vst", nfeatures = 1000)
sce.all <- ScaleData(sce.all)
sce.all <- RunPCA(sce.all, features = VariableFeatures(sce.all))
sce.all <- RunHarmony(sce.all, "orig.ident")
p1 <- ElbowPlot(sce.all, ndims = 30)
ggsave(filename = "0.pbmc_elbowplot.png", plot = p1,width = 8,height = 8,dpi = 300)
sce.all <- FindNeighbors(sce.all, reduction = "harmony", dims = 1:24)
sce.all <- FindClusters(sce.all, resolution = 0.5)
sce.all <- RunUMAP(sce.all, dims = 1:24, reduction = "harmony")
sce.all <- RunTSNE(sce.all, dims = 1:24, reduction = "harmony")
p2 <- DimPlot(sce.all, reduction = "umap", label = T, repel = T, label.size = 5)
ggsave(filename = "0.pbmc_umap_in_cluster_hgv1000_pca24_r0.5.png", plot = p2,width = 8,height = 8,dpi = 300)
saveRDS(sce.all, file = "pbmc.cluster.hgv1000.pca24.r0.5.sce.all")六、Marker基因的选取及注释分群
上一步已经确定了聚类多少个cluster,但这些cluster是什么细胞类型我们并不知道,这时候需要生物学背景通过一些网站去找Marker基因,来标识细胞类型,例如celltaxonmy和cellMarker,也可以通过最新的一些文献去选取常用的Marker基因,先将细胞大类群注释出来,例如免疫细胞、上皮、内皮等。PS:这里可以再额外多了解一些不同组织常见的细胞类型。最好的方案是去找文献看已有的文章是怎么对某一个组织进行大类分群的来确定,相应的marker基因文章也会给出,可以多综合几篇文章,把marker基因都扒取下来作为候选。例例如seurat官网给的,当然肯定是不够的。

等到所有的cluster都能确定类型后就可以开始命名了,当然肯定有一些cluster没办法确定类型,一个方案是继续找marker,总有能标识他的,另一个方案是把它并到umap图里更近的cluser,还有一种可能是这群是多胞,那自然就不需要这群。下面的代码是常规的细胞命名的代码,当cluster群多了的时候容易看花眼并不好用,但是初学者看这个会更容易。另一种方案是通过分别构造向量命名同一细胞类型,下面也给出了。
###cluster从0到最后一个一一对应。
new.cluster.ids <- c("Naive CD4 T", "CD14+ Mono", "Memory CD4 T", "B", "CD8 T", "FCGR3A+ Mono",
"NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
###当有多个cluster是同一类群时,特别是我们在大类注释时例如将T/NK合并注释大类,通常用下面的方式更方便。
###例如有0-18个cluster
###下面的命名不具有意义,只是举例
celltype = data.frame(ClusterID=0:18,
celltype= 0:18)
celltype[celltype$ClusterID %in% c(1,3,4,5,7,15),2]='T/NK'
celltype[celltype$ClusterID %in% c(6),2]='B'
celltype[celltype$ClusterID %in% c(8,18),2]='MD'
celltype[celltype$ClusterID %in% c(13),2]='Neutrophils'
celltype[celltype$ClusterID %in% c(0,2,9,12,16),2]='DC'
celltype[celltype$ClusterID %in% c(11),2]='Endo'
celltype[celltype$ClusterID %in% c(14),2]='FL'
celltype[celltype$ClusterID %in% c(10),2]='Platelet'
celltype[celltype$ClusterID %in% c(17),2]='Plasma'
new.cluster.ids=celltype$celltype
names(new.cluster.ids) <- levels(sce.all)
sce.all <- RenameIdents(sce.all, new.cluster.ids)
sce.all$cellType=Idents(sce.all)最终图呈现
#############################最终图呈现调整##################################
p4 <- DimPlot(sce.all, reduction = "umap", label = TRUE, pt.size = 0.5, group.by = "ident")
ggsave(filename = "1.pbmc_umap_in_celltype_hgv1000_pca24_r0.5.png", plot = p4,width = 8,height = 8,dpi = 300)
p5 <- DotPlot(sce.all, features = markers2, dot.scale = 8, group.by = 'cellType') + RotatedAxis() +
theme(
panel.background = element_blank(), # 移除面板背景
plot.background = element_rect(fill = "white"), # 确保绘图区白色
axis.line = element_line(color = "black"), # 保留坐标轴线
panel.grid = element_blank() # 移除网格线
)
ggsave(filename = "1.pbmc_marker2_in_celltype_hgv1000_pca24_r0.5.png", plot = p5,width = 16,height = 8,dpi = 300)
###保存数据
saveRDS(sce.all, file = "pbmc.hgv1000.pca24.r0.5.cellTypeAssigned.sce.all")
###############################################################################七、关于亚群的细分
亚群细分是提取某一个细胞大类重新进行亚型分类,这样的目的是为了使内部分的更准确。
以下以T/NK免疫细胞亚群分型为例。后面的过程就是贴标签,重复先前的过程。这里需要注意nfeature、dims参数的变化,缩减为了先前的一半,对于不同的数据集也许不太一样,但是对于一个亚群内部而言肯定不需要先前那么多的高变基因了,更多的高变进来会形成噪音干扰导致区分度不高。对于其他的亚群也是一样的做法,这样就可以完成亚群的细分了,最后汇总所有有用的marker,那就可以再整体umap上标记所有的细胞类型及亚型啦。
###############################################################################
#############################区分TNK亚型######################################
###############################################################################
TNK_markers <- c( "NKG7","GNLY","NCAM1", #NK cell
"CD8A","CD96","CD8B",#"GZMH",#T/CD8+T
"IL7R", "CD4" #T/CD4+ T
#以下略
)
sce.all <- readRDS("pbmc.hgv1000.pca24.r0.5.cellTypeAssigned.sce.all")
sce.all <- subset(sce.all, idents = "T/NK")
sce.all <- NormalizeData(sce.all, normalization.method = "LogNormalize", scale.factor = 10000)
sce.all <- FindVariableFeatures(sce.all, selection.method = "vst", nfeatures = 500)
sce.all <- ScaleData(sce.all)
sce.all <- RunPCA(sce.all, features = VariableFeatures(sce.all))
sce.all <- RunHarmony(sce.all, "orig.ident")
p6 <- ElbowPlot(sce.all, ndims = 20)
ggsave(filename = "2.pbmc_TNK_hgv500_elbowplot.png", plot = p6,width = 8,height = 8,dpi = 300)
sce.all <- FindNeighbors(sce.all, reduction = "harmony", dims = 1:12)
sce.all <- FindClusters(sce.all, resolution = 0.5)
sce.all <- RunUMAP(sce.all, dims = 1:12, reduction = "harmony")
sce.all <- RunTSNE(sce.all, dims = 1:12, reduction = "harmony")
p7 <- DimPlot(sce.all, reduction = "umap", label = T, repel = T, label.size = 5)
ggsave(filename = "2.pbmc_TNK_umap_in_cluster_hgv500_pca12_r0.5.png", plot = p7,width = 8,height = 8,dpi = 300)
p8 <- DotPlot(sce.all, features = TNK_markers, dot.scale = 8) + RotatedAxis()
ggsave(filename = "2.pbmc_TNK_markers_in_cluster_hgv500_pca12_r0.5.png", plot = p8,width = 8,height = 8,dpi = 300)
############################################################################
#############################区分TNK亚型######################################
############################################################################八、后记
关于单细胞的学习,向师弟以及兄弟官印和室友津津请教了很多,在此感谢他们的耐心解答。
同时也感谢老板给我这个机会学习尝试一些新的东西。
笔者自知对于单细胞分析的认识还比较浅薄,只能当是记载心路历程和踩过的一些坑,供参考,持续学习,互勉。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号