

Nature子刊共线性作图复现
文献如下,今天再读文献时,发现这篇文章是给出了原始数据甚至作图代码的,于是前来follow复现一下其中的图4A。主要是学一下数据处理及格式转换部分。
Belser C, Baurens FC, Noel B, Martin G, Cruaud C, Istace B, Yahiaoui N, Labadie K, Hřibová E, Doležel J, Lemainque A, Wincker P, D'Hont A, Aury JM. Telomere-to-telomere gapless chromosomes of banana using nanopore sequencing. Commun Biol. 2021 Sep 7;4(1):1047. doi: 10.1038/s42003-021-02559-3. PMID: 34493830; PMCID: PMC8423783.
###jcvi安装及教程
https://github.com/tanghaibao/jcvi
https://github.com/tanghaibao/jcvi/wiki/MCscan-(Python-version)
https://www.cnblogs.com/liuxinxin21d/articles/16792258.html
### 作图代码:
https://github.com/institut-de-genomique/Pahang-associated-data/tree/main/Figure4A
### 数据来源:
Belser C, Baurens FC, Noel B, Martin G, Cruaud C, Istace B, Yahiaoui N, Labadie K, Hřibová E, Doležel J, Lemainque A, Wincker P, D'Hont A, Aury JM. Telomere-to-telomere gapless chromosomes of banana using nanopore sequencing. Commun Biol. 2021 Sep 7;4(1):1047. doi: 10.1038/s42003-021-02559-3. PMID: 34493830; PMCID: PMC8423783.
https://github.com/institut-de-genomique/Pahang-associated-data/tree/main/Figure4A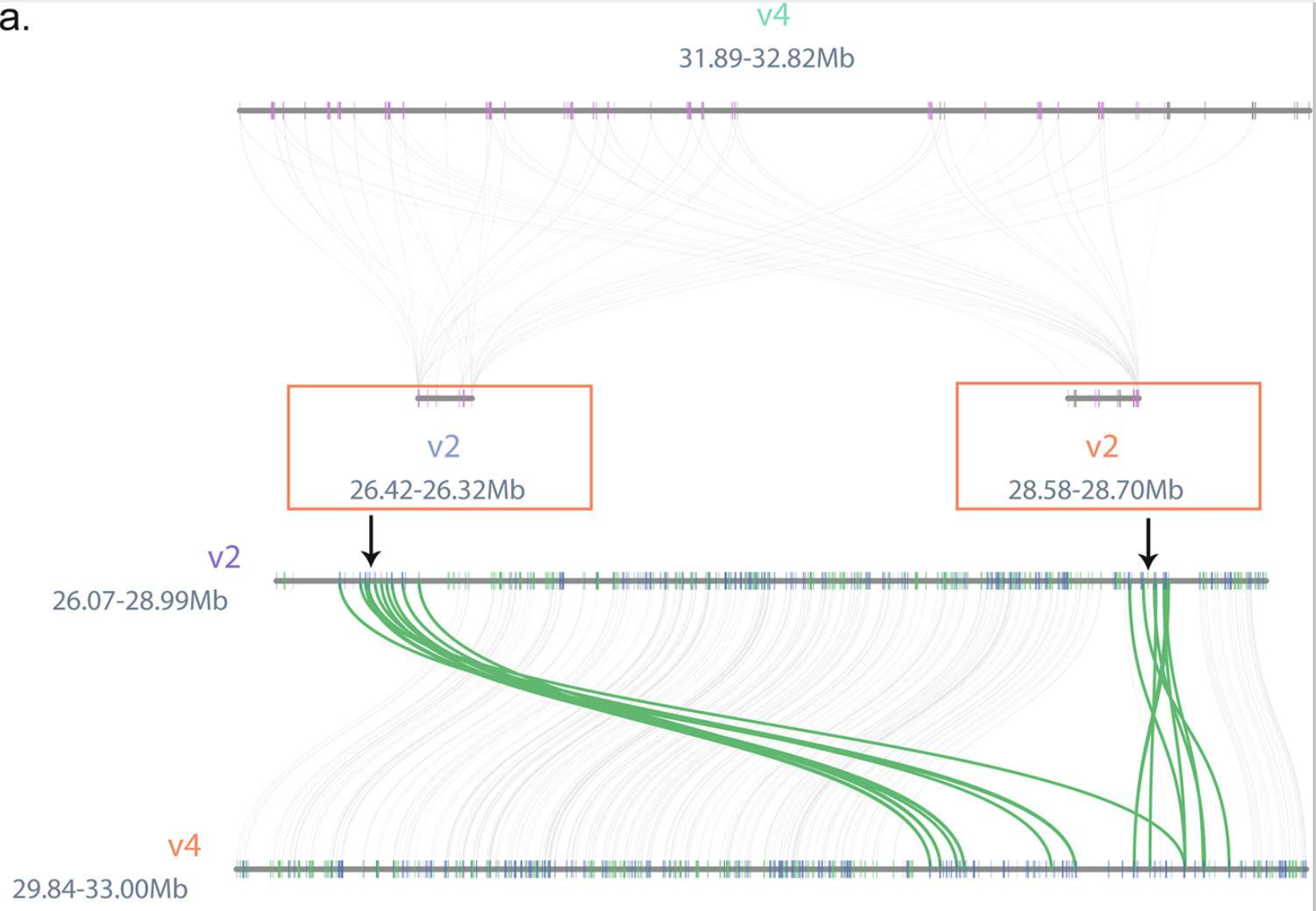
一、绘制上半部分
V2版本的萜类化合物合成基因定位到了两个分开的区域,V4版本由于基因组拼接质量更优,相关基因定位于一个密集区域。因此这部分作图分成主要是想看V2与V4间的萜类化合物合成基因的共线性情况,类比三物种共线性作图分析。
STEP 01 提取目标TDGs的gff文件
##00 prepare data(v4版本BGH可以找到,v2版本的需要跑一遍文章里gene prediction部分的流程才能获得)
Musa_acuminata_pahang_v4.gff
musa_acuminata_v2.gff3
DH-Pahang.4.2.fasta
musa_acuminata_v2_pseudochromosome.fna
##01 extraction of the terpene synthase genes in the V4 annotation gff file
for i in Macma4_07_g21890.1 Macma4_07_g21860.1 Macma4_07_g21850.1 Macma4_07_g21820.1 Macma4_07_g21830.1 Macma4_07_g21940.1 Macma4_07_g21790.1 Macma4_07_g21770.1 Macma4_07_g21780.1 Macma4_07_g21690.1 Macma4_07_g21800.1 Macma4_07_g21870.1 Macma4_07_g21930.1 Macma4_07_g21900.1 Macma4_07_g21840.1 Macma4_07_g21700.1 Macma4_07_g21610.1 Macma4_07_g21550.1 Macma4_07_g21540.1 Macma4_07_g21620.1 Macma4_07_g21580.1 Macma4_07_g21480.1 Macma4_07_g21440.1 Macma4_07_g21420.1 Macma4_07_g21500.1 Macma4_07_g21530.1 Macma4_07_g21570.1 Macma4_07_g21430.1 Macma4_07_g21590.1 Macma4_07_g21460.1 Macma4_07_g21450.1 Macma4_07_g21660.1 Macma4_07_g21650.1 Macma4_07_g21730.1 Macma4_07_g21560.1 Macma4_07_g21640.1 Macma4_07_g21740.1 Macma4_07_g21910.1; do cat Musa_acuminata_pahang_v4.gff3 | grep $i >> TDGs_V4.gff; done;
##02 extraction of the terpene synthase genes in the V2 annotation gff file : one for the left cluster and one for the right cluster :
for i in Ma07_t18590.1 Ma07_t18650.1 Ma07_t18570.1 Ma07_t18560.1 Ma07_t18640.1 Ma07_t18610.1,Ma07_t18580.1,Ma07_t18680.1 Ma07_t18630.1 Ma07_t18540.1 Ma07_t18620.1; do cat musa_acuminata_v2.gff3 | grep $i >> TDGs_V2_left.gff; done;
for i in Ma07_t20710.1 Ma07_t20740.1 Ma07_t20750.1 Ma07_t20660.1 Ma07_t20700.1 Ma07_t20680.1 Ma07_t20650.1 Ma07_t20720.1 Ma07_t20730.1; do cat musa_acuminata_v2.gff3 | grep $i >> TDGs_V2_right.gff; done;
##至此获得jcvi输入数据如下
TDGs_V4.gff
TDGs_V2_left.gff
TDGs_V2_right.gffSTEP 02 准备jcvi输入文件
##00 prepare data
TDGs_V4.gff
TDGs_V2_left.gff
TDGs_V2_right.gff
DH-Pahang.4.2.fasta
musa_acuminata_v2_pseudochromosome.fna
##01 GFF2BED
python -m jcvi.formats.gff bed --type=CDS --key=Parent TDGs_V4.gff -o v4.bed
python -m jcvi.formats.gff bed --type=CDS --key=Parent TDGs_V2_left.gff -o v2_left.bed
python -m jcvi.formats.gff bed --type=CDS --key=Parent TDGs_V2_right.gff -o v2_right.bed
cat v4.bed | awk '{print $1"\t"$2"\t"$3"\t""Parent="$4"\t"$5"\t"$6}' > temp
mv temp v4.bed
cat v2_left.bed | awk '{print $1"\t"$2"\t"$3"\t""Parent="$4"\t"$5"\t"$6}' > temp
mv temp v2_left.bed
cat v2_right.bed | awk '{print $1"\t"$2"\t"$3"\t""Parent="$4"\t"$5"\t"$6}' > temp
mv temp v2_right.bed
##02 提取TDGs目标基因对应的序列
# 提取目标序列(这里用到的gff2fasta没有找到是哪个软件的工具,跳过这一步)
gff2fasta -i TDGs_V4.gff -b DH-Pahang.4.2.fasta -o v4_TDG_CDS.fasta -type CDS
gff2fasta -i TDGs_V2_left.gff -b musa_acuminata_v2_pseudochromosome.fna -o v2_TDG_CDS_left.fasta -type CDS
gff2fasta -i TDGs_V2_right.gff -b musa_acuminata_v2_pseudochromosome.fna -o v2_TDG_CDS_right.fasta -type CDS
# 更改fasta序列文件格式,使其符合jcvi输入要求(作者的包里给出了下列fasta文件,直接从这一步开始)
python -m jcvi.formats.fasta format v4_TDG_CDS.fasta v4.cds #(v4_TDG_CDS.fasta也没有给,跳过这一步,直接用给出的结果v4.cds)
python -m jcvi.formats.fasta format v2_TDG_CDS_left.fasta v2_left.cds
python -m jcvi.formats.fasta format v2_TDG_CDS_right.fasta v2_right.cdsSTEP 03 JCVI作图从此处开始:上半部分(实际上就是JCVI三物种作图流程来展示)
###这部分的逻辑类似于JCVI官网的三物种共线性分析
# 构建v2_left与v4间的synteny blocks
python -m jcvi.compara.catalog ortholog v4 v2_left --no_strip_names --notex
python -m jcvi.compara.synteny screen --minspan=1 --simple v4.v2_left.anchors v4.v2_left.anchors.new
python -m jcvi.compara.synteny mcscan v4.bed v4.v2_left.lifted.anchors --iter=1 -o v4.v2_left.i1.blocks
# 构建v2_right与v4间的synteny blocks
python -m jcvi.compara.catalog ortholog v4 v2_right --no_strip_names --notex
python -m jcvi.compara.synteny screen --minspan=1 --simple v4.v2_right.anchors v4.v2_right.anchors.new
python -m jcvi.compara.synteny mcscan v4.bed v4.v2_right.lifted.anchors --iter=1 -o v4.v2_right.i1.blocks
# Combining the blocks to one blocks
python -m jcvi.formats.base join v4.v2_right.i1.blocks v4.v2_left.i1.blocks --noheader | cut -f1,2,4,6 > cut.blocks
# 构建cut.layout
# x, y, rotation, ha, va, color, ratio, label
0.5, 0.6, 0, center, top, , 1, v2
0.3, 0.4, 0, center, bottom, , .5, v2
0.7, 0.4, 0, center, bottom, , .5, v4
# edges
e, 0, 1
e, 0, 2
# 合并*.bed文件
cat v4.bed v2_right.bed v2_left.bed > right_left_v4.bed
# 准备好cut.blocks right_left_v4.bed cut.layout后一键作图
python -m jcvi.graphics.synteny cut.blocks right_left_v4.bed cut.layout --glyphcolor=orthogroup --notex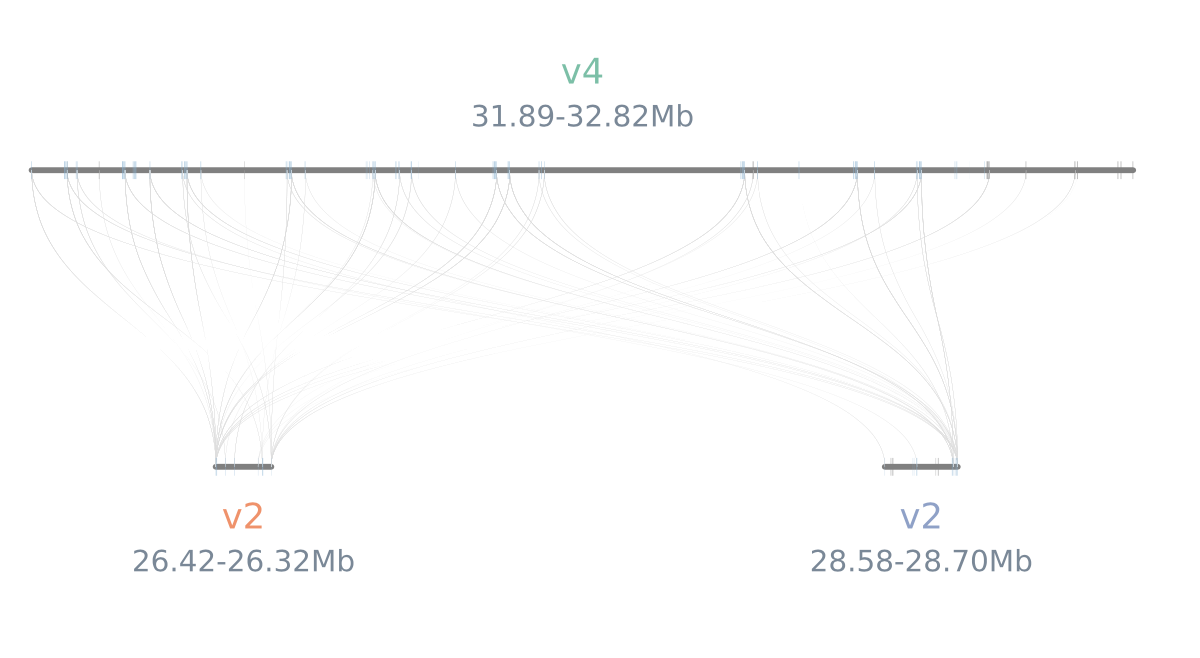
二、绘制下半部分
该部分作图是针对V2与V4版本的7号染色体上包含萜类化合物合成基因区域部分的共线性分析。
jcvi的流程部分大抵没什么变化,主要是学习一下针对某个特定区域去做共线性分析的数据处理流程。
PS: 这部分作者给的流程有问题,没法按照流程出最终的结果。需要另外修改。
STEP 01 数据准备及格式转换
## 定位V4版本7号染色体的29.60Mb至33.00Mb区域的行输出为新的gff
cat Musa_acuminata_pahang_v4.gff3 | grep chr07 | awk '{if($4>29600000 && $5<33000000){print $0}}' > expanded_chr07_region_v4.gff
## 定位V2版本7号染色体的26.00Mb至29.00Mb区域的行输出为新的gff
cat musa_acuminata_v2.gff3 | grep chr07 | awk '{if($4>26000000 && $5<29000000){print $0}}' > expanded_chr07_region_v2.gff
# GFF2BED file and rename them.
python -m jcvi.formats.gff bed --type=CDS --key=Parent expanded_chr07_region_v2.gff -o expanded_v2.bed
python -m jcvi.formats.gff bed --type=CDS --key=Parent expanded_chr07_region_v4.gff -o expanded_v4.bed
cat expanded_v4.bed | awk '{print $1"\t"$2"\t"$3"\t""Parent="$4"\t"$5"\t"$6}' > temp
mv temp expanded_v4.bed
cat expanded_v2.bed | awk '{print $1"\t"$2"\t"$3"\t""Parent="$4"\t"$5"\t"$6}' > temp
mv temp expanded_v2.bed
# 提取该区域对应的cds序列
gff2fasta -i expanded_chr07_region_v4.gff -b DH-Pahang.4.2.fasta -o expanded_v4_TDG_CDS.fasta -type CDS
gff2fasta -i expanded_chr07_region_v2.gff -b musa_acuminata_v2_pseudochromosome.fna -o expanded_v2_TDG_CDS.fasta -type CDS
# 更改提取出来序列的格式
python -m jcvi.formats.fasta format expanded_v2_TDG_CDS.fasta expanded_v2.cds
python -m jcvi.formats.fasta format expanded_v4_TDG_CDS.fasta expanded_v4.cds
## 先看一眼数据,现在应该有了如下数据
ls *.cds *.bed
expanded_v2.cds
expanded_v4.cds
expanded_v2.bed
expanded_v4.bedSTEP02 JCVI作图从此处开始
# snyteny搜索
python -m jcvi.compara.catalog ortholog expanded_v2 expanded_v4 --no_strip_names --notex
# *.simple文件生成
python -m jcvi.compara.synteny screen --minspan=1 --simple expanded_v2.expanded_v4.anchors expanded_v2.expanded_v4.anchors.new
# refine *.blocks
python -m jcvi.compara.synteny mcscan expanded_v2.bed expanded_v2.expanded_v4.lifted.anchors --iter=1 -o expanded_v2.expanded_v4.i1.blocks
# 合并*.bed文件
cat expanded_v2.bed expanded_v4.bed > expanded_v2_v4.bed
# 配置layout
more expanded_blocks.layout
# x, y, rotation, ha, va, color, ratio, label
0.5, 0.6, 0, left, center, m, 1, v2
0.5, 0.4, 0, left, center, #fc8d62, 1, v4
# edges
e, 0, 1
# 准备好*.blocks、*.bed以及*.layout文件后一键绘图
python -m jcvi.graphics.synteny expanded_v2.expanded_v4.i1.blocks expanded_v2_v4.bed expanded_blocks.layout --notexSTEP03 JCVI作图优化:修改blocks文件,高亮突出v2中的基因
# 首先整理出需要高亮的基因id:将作者给的v2基因id整理成一行,以空格分割的文件highlight.gene.id, 文件内容如下
Ma07_t18590.1 Ma07_t18650.1 Ma07_t18570.1 Ma07_t18560.1 Ma07_t18640.1 Ma07_t18610.1 Ma07_t18580.1 Ma07_t18680.1 Ma07_t18630.1 Ma07_t18540.1 Ma07_t18620.1 Ma07_t20710.1 Ma07_t20740.1 Ma07_t20750.1 Ma07_t20660.1 Ma07_t20700.1 Ma07_t20680.1 Ma07_t20650.1 Ma07_t20720.1 Ma07_t20730.1# 手写py脚本,vim doHighlightBlocks.py
#打开需要高亮的gene.id文件,准备id列表用于查询
f1 = open(r'/home/liuxin/Figure4A/Expanded_region/test2/highlight.gene.id','r')
f1line = f1.readline()
id = f1line.split(' ')
for i in id:
print (i)
#批量高亮blocks文件中与目标gene.id一致的行
with open(r'/home/liuxin/Figure4A/Expanded_region/test2/expanded_v2.expanded_v4.i1.blocks','r') as raw_file:
while True:
line = raw_file.readline()
if not line:
break
else:
if line.startswith('#'):
continue
cur_line = line.split('\t')
blocksline_id = cur_line[0].split('=')[-1]
if blocksline_id in id:
#以r*开头的行将标记为红色
new_line = 'r*'+cur_line[0]+'\t'+cur_line[1]
print(new_line)
with open('/home/liuxin/Figure4A/Expanded_region/test2/highlight.gene.blocks','a') as save_file:
save_file.write(new_line)
else:
print(line)
with open('/home/liuxin/Figure4A/Expanded_region/test2/highlight.gene.blocks','a') as save_file:
save_file.write(line)# 执行脚本文件
python doHighlightBlocks.py# 得到高亮突出过后的highlight.gene.blocks文件,重新绘图
python -m jcvi.graphics.synteny highlight.gene.blocks expanded_v2_v4.bed expanded_blocks.layout --notex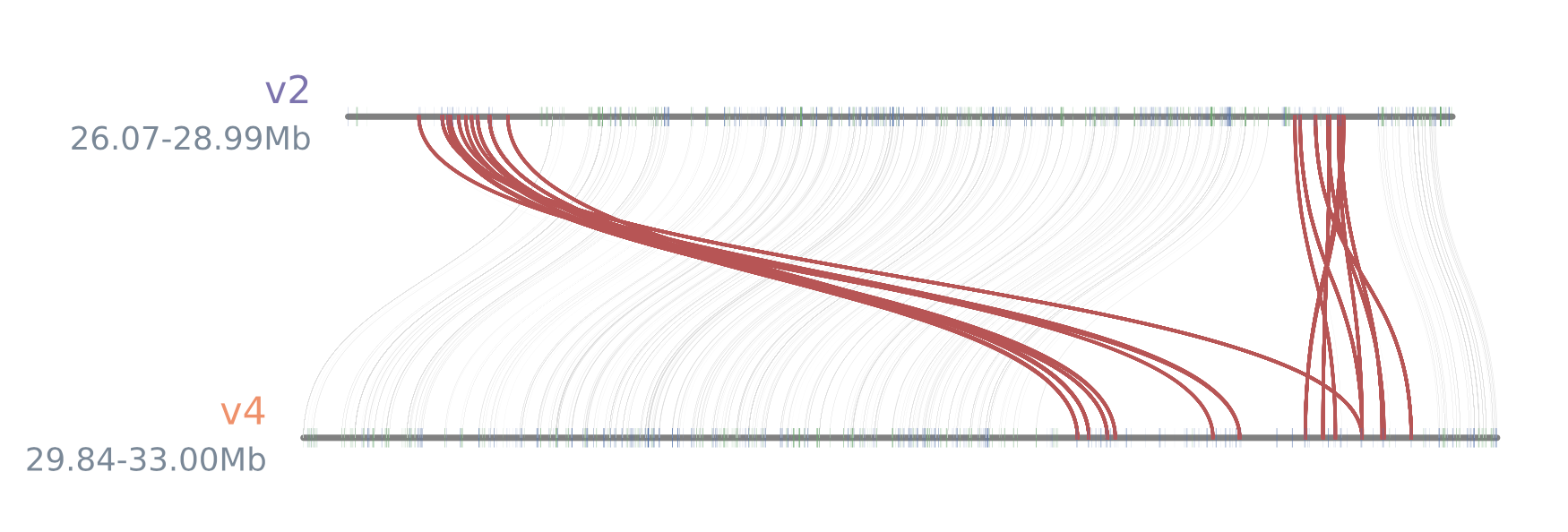
三、讨论
将上下两部分导入AI拼接在一起,稍做修改即可得到与文章一致的图。主要还是学习前边数据处理与转换部分,jcvi的作图部分可以说是很简单了。但是批量高亮基因也是个麻烦事,手动去调整不现实,文中的脚本给出了批量高亮标注blocks的方法,本来打算用awk去实现,想来想去还是python更容易。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号