
Linux 中如何将fasta文件的每一个scaffold的碱基转换成一行显示
001、awk
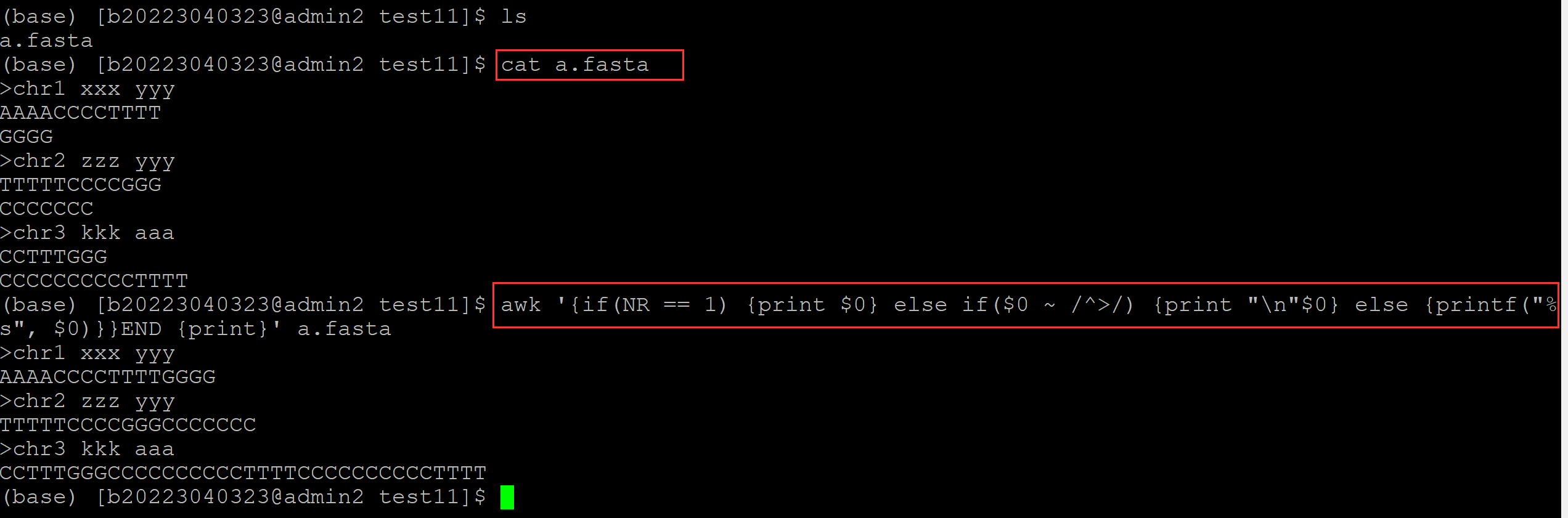
(base) [b20223040323@admin2 test11]$ ls a.fasta (base) [b20223040323@admin2 test11]$ cat a.fasta ## 测试fasta文件 >chr1 xxx yyy AAAACCCCTTTT GGGG >chr2 zzz yyy TTTTTCCCCGGG CCCCCCC >chr3 kkk aaa CCTTTGGG CCCCCCCCCCTTTT (base) [b20223040323@admin2 test11]$ awk '{if(NR == 1) {print $0} else if($0 ~ /^>/) {print "\n"$0} else {printf("%s", $0)}}END {print}' a.fasta ## awk实现 >chr1 xxx yyy AAAACCCCTTTTGGGG >chr2 zzz yyy TTTTTCCCCGGGCCCCCCC >chr3 kkk aaa CCTTTGGGCCCCCCCCCCTTTTCCCCCCCCCCTTTT
002、seqkit实现
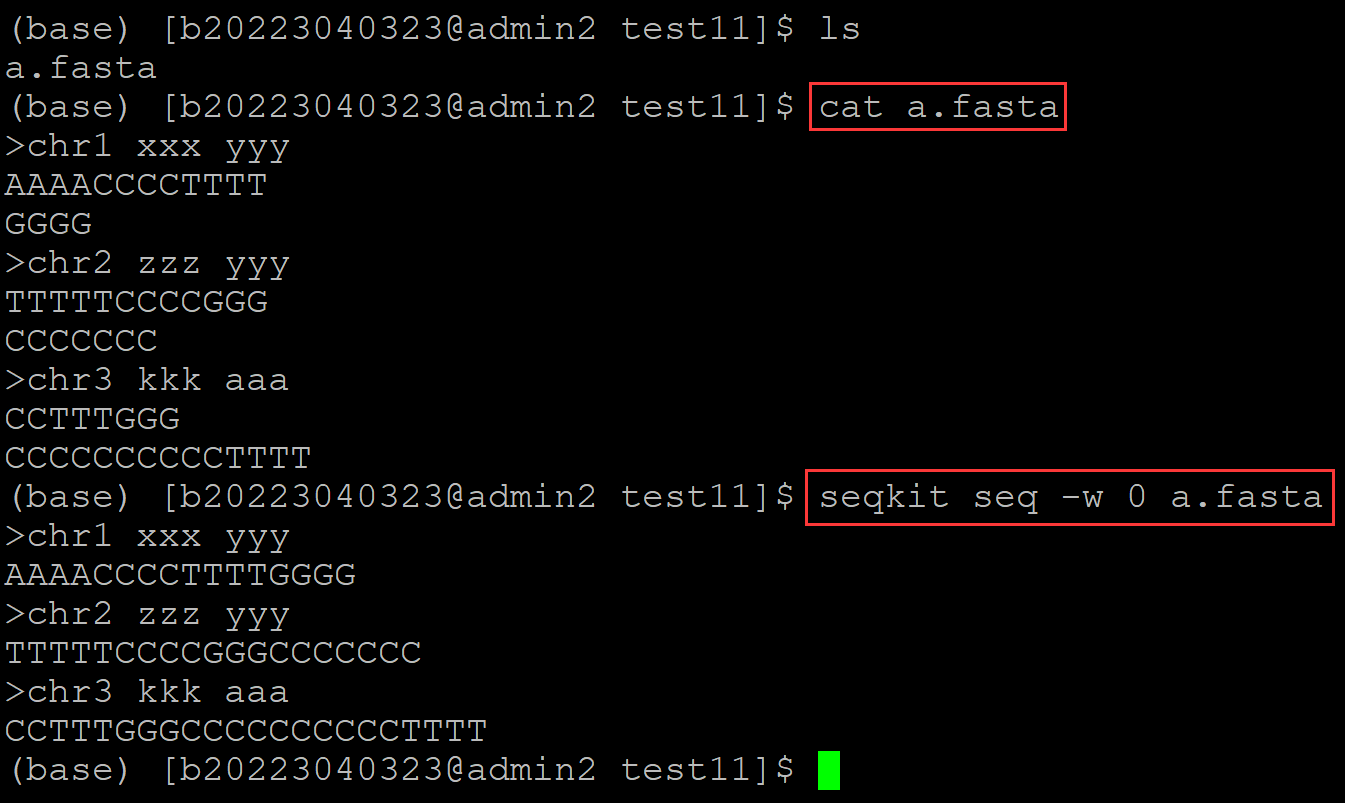
(base) [b20223040323@admin2 test11]$ ls a.fasta (base) [b20223040323@admin2 test11]$ cat a.fasta ## 测试fasta文件 >chr1 xxx yyy AAAACCCCTTTT GGGG >chr2 zzz yyy TTTTTCCCCGGG CCCCCCC >chr3 kkk aaa CCTTTGGG CCCCCCCCCCTTTT (base) [b20223040323@admin2 test11]$ seqkit seq -w 0 a.fasta ## seqkit 实现 >chr1 xxx yyy AAAACCCCTTTTGGGG >chr2 zzz yyy TTTTTCCCCGGGCCCCCCC >chr3 kkk aaa CCTTTGGGCCCCCCCCCCTTTT
。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号