

seqkit软件根据染色体名称从fasta文件中批量提取数据
001、批量提取,将染色体名称写入到list文件
[root@pc1 test1]# ls a.fa chr.list [root@pc1 test1]# cat a.fa ## 测试fasta >chr1 tttcccggg >chr2 tttggg ccc >chr3 cccttt >chr4 aaaaattt [root@pc1 test1]# cat chr.list ## 染色体列表 chr2 chr4 [root@pc1 test1]# seqkit -w 8 grep -f chr.list a.fa > result.fa ## -w指定每行输出的碱基数目 [INFO] 2 patterns loaded from file [root@pc1 test1]# cat result.fa ## 结果文件 >chr2 tttgggcc c >chr4 aaaaattt
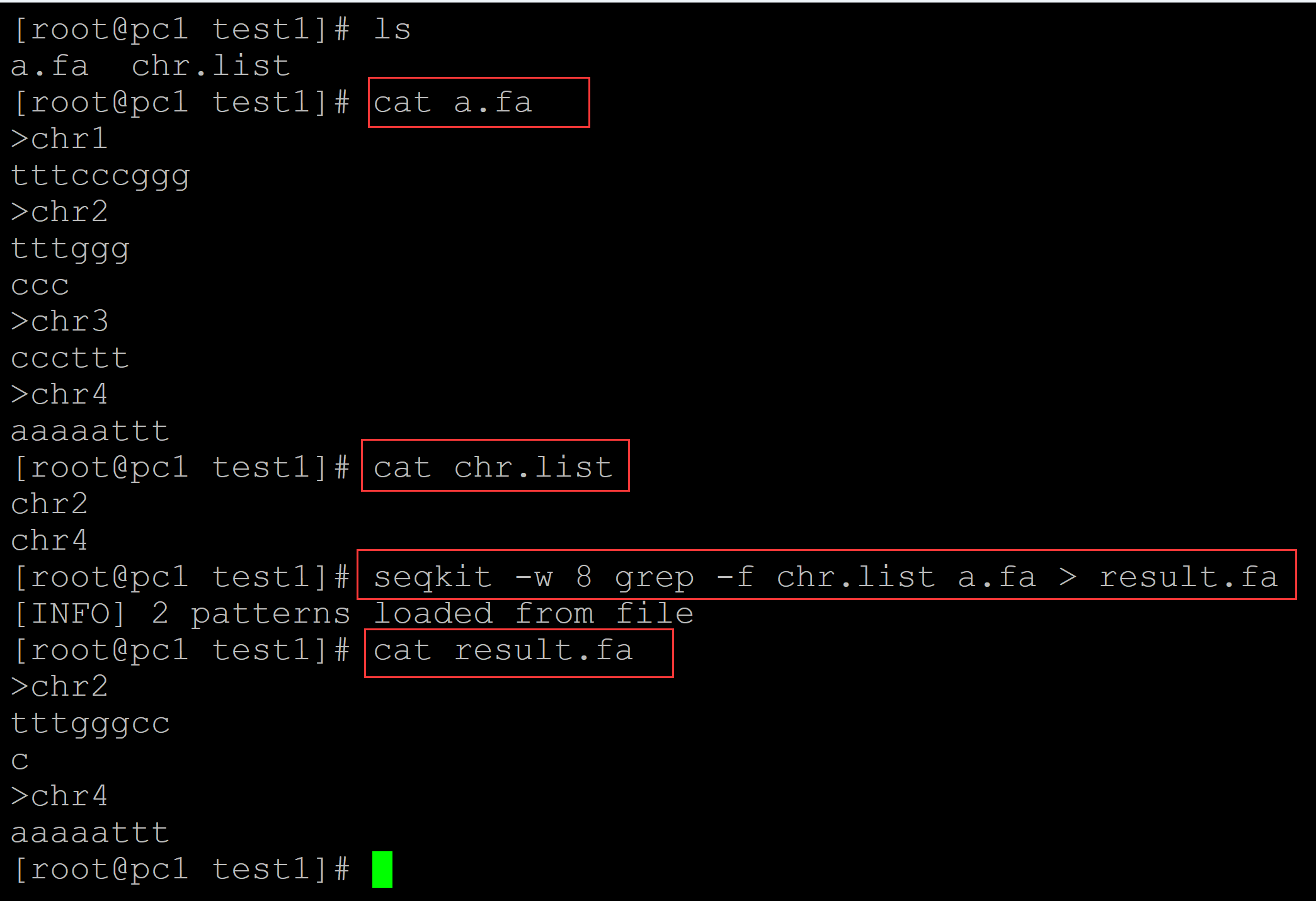
。
002、根据单条染色体的名称提取
seqkit grep -p "NC_056054.1" GCF_016772045.1_ARS-UI_Ramb_v2.0_genomic.fna
。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号