

bam文件去重复
建库过程PCA扩增过程中引入重复序列,会对变异检测结果产生影响,重复的DNA片段会比对到参考基因组的相同位置,根据这一特点来进行去重复。
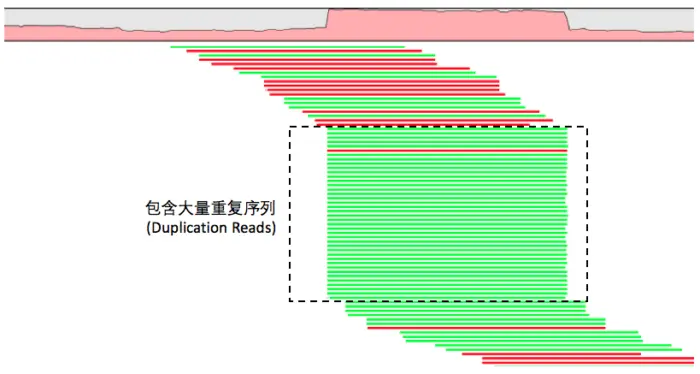
001、gatk(picard标记重复)
gatk MarkDuplicates -I sample01.sorted.bam -O sample01.sorted.markdup.bam -M sample01.sorted.markdup_metrics.txt
002、samtools
samtools sort -n xxx.bam -o xxx.sort.bam ## step1,按read name排序 samtools fixmate -m xxx.sort.bam xxx.fixmate.bam ## step2 samtools sort xxx.fixmate.bam -o xxx.positionsort.bam ## step3,按position排序 samtools markdup -r xxx.positionsort.bam xxx.markdup.bam ## step4,加上-r就会直接去掉重复序列
## 或者合并命令: samtools sort -n xxx.bam | samtools fixmate -m | samtools sort | samtools markdup -r > xxx.markdup.bam
参考:
01、https://www.jianshu.com/p/8cdbb89530c6?utm_campaign=maleskine&utm_content=note&utm_medium=seo_notes
02、https://www.cnblogs.com/bio-mary/p/12053346.html
03、https://www.jianshu.com/p/e20a3b73dcd0
。
.

 浙公网安备 33010602011771号
浙公网安备 33010602011771号