

Linux 中 shell 脚本实现根据gff 计算每一个基因的最长转录本
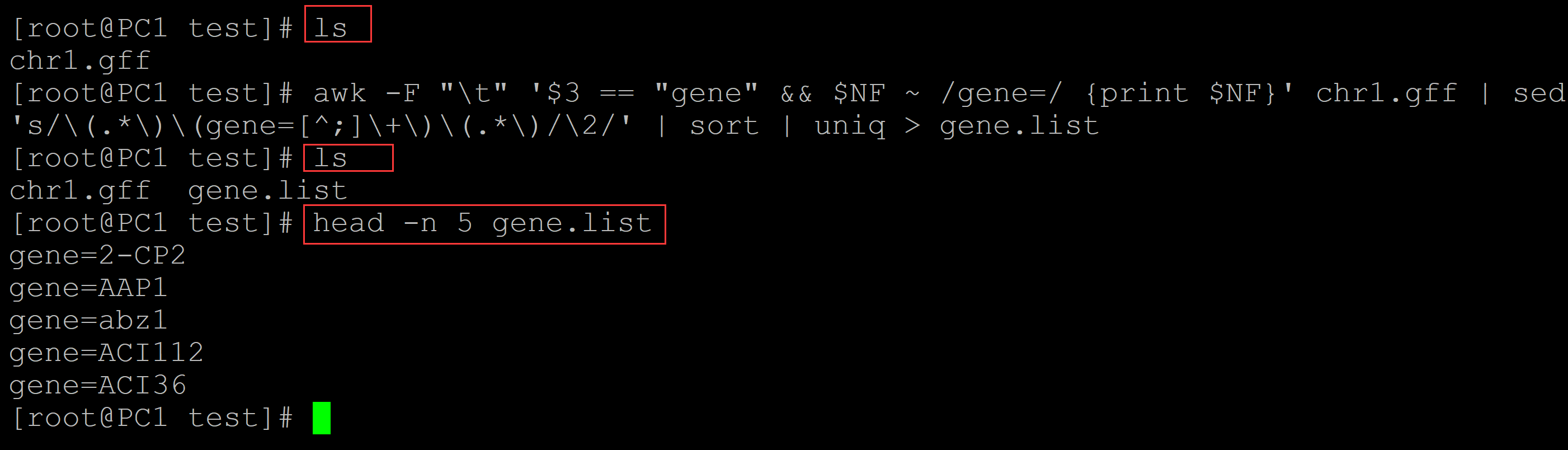
001、生成基因名称的列表
awk -F "\t" '$3 == "gene" && $NF ~ /gene=/ {print $NF}' chr1.gff | sed 's/\(.*\)\(gene=[^;]\+\)\(.*\)/\2/' | sort | uniq > gene.list
002、计算最长转录本
a、脚本文件
#!/bin/bash num=0 for i in $(cat gene.list) do let num=$num+1 mRNA=$(grep "$i;" chr1.gff | awk '$3 ~ /^mRNA$/' | wc -l) exon=$(grep "$i;" chr1.gff | awk '$3 ~ /^exon$/' | wc -l) if [ $exon -eq 0 ] then echo "$i has no exon" exit fi if [ $mRNA -gt 1 ] then grep "$i;" chr1.gff | awk '$3 ~ /^mRNA$|^exon$/' | cut -f 3-5 | awk 'BEGIN{a=0}{if($1 ~ /^mRNA$/){a++}; if($1 ~ /^exon$/) {print "exon"a, $2, $3}}' | awk '{ay[$1] += ($3 - $2 + 1)} END {for(j in ay) print j, ay[j]}' | awk -v sam=$i '{if(NR == 1) {max = $2} else if($2 > max) {max = $2}} END {print sam, max}' >> result.txt else grep "$i;" chr1.gff | awk -v sam=$i '$3 ~ /^exon$/ {sum += ($5 - $4 + 1)} END {print sam, sum}' >> result.txt fi echo $num done done
b、执行程序
[root@PC1 test]# bash record.sh
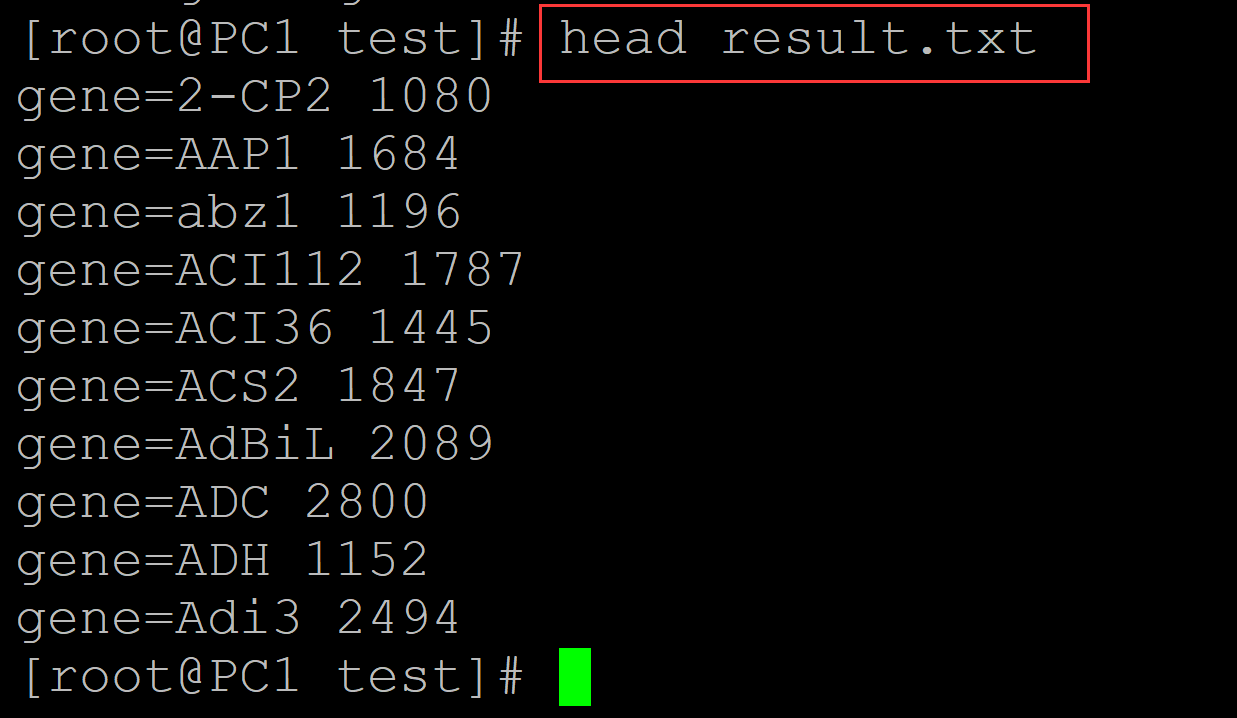
c、结果文件:
[root@PC1 test]# head result.txt gene=2-CP2 1080 gene=AAP1 1684 gene=abz1 1196 gene=ACI112 1787 gene=ACI36 1445 gene=ACS2 1847 gene=AdBiL 2089 gene=ADC 2800 gene=ADH 1152 gene=Adi3 2494

 浙公网安备 33010602011771号
浙公网安备 33010602011771号