
7Free Energies: 7.1 MM-PBSA
2022.11.24更新:使用MMPBSA计算(小分子) 和 (蛋白+其它小分子)的结合自由能
MMPBSA是可以做的,实践上发现只能使用MMPBSA.py,或者MMPBA.py.MPI调用一个线程,不能并行。
例子:计算泛醇分子从LH1中解离后,在膜内的结合自由能。如果使用常规的MMPBSA,即只考虑泛醇分子和蛋白质,则计算的结合自由能接近0kcal/mol。 这是不合理的,因为常规的MMPBSA计算都只考虑蛋白质和小分子,忽略了醌分子与LH1外部的膜之间的范德华和静电相互作用,即使使用隐式膜模型,也只是解决了和膜分子之间的静电相互作用,所以醌在外部膜中的自由能计算值一直接近0。(而醌分子在外部膜内更可能是通过范德华相互作用而结合)
知道了这一点,可以使用“非常规”的方法,就是在MMPBSA计算的时候,将体系中除了水分子之外的所有原子都考虑为显式的,虽然计算可能会非常耗时,但这样获得的结果在这种情况下是更准确的。
2021.12.1更新 :关于隐式膜MMPBSA
参考https://www.cnblogs.com/jszd/p/11215880.html
7.1MM-PBSA
在本教程中,我们将使用 MM-PBSA 方法计算两种蛋白质结合的结合自由能。
- 介绍1
理想状态下,计算结合自由能:
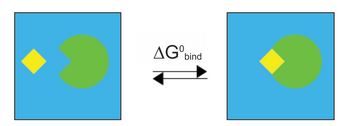
然而,在这些溶剂化状态的这种模拟中,大部分能量贡献将来自溶剂 - 溶剂相互作用,并且总能量的波动将比结合能大一个数量级。 因此,计算将花费过多的时间来收敛。 因此,更有效的方法是根据以下热力学循环划分计算:
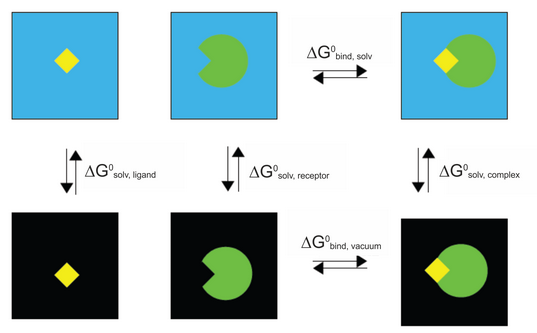
显然,从该图中可以计算出结合自由能:
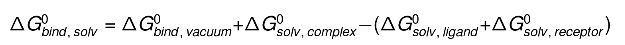
溶剂化自由能是通过求解三种状态中每一种的线性泊松玻尔兹曼方程或广义玻恩方程来计算的(这给出了对溶剂化自由能的静电贡献)并添加了疏水贡献的经验项:
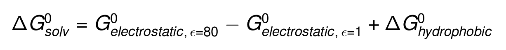
通过计算受体和配体之间的平均相互作用能并在必要/需要时考虑结合时的熵变化来获得 delta-G vacuum:
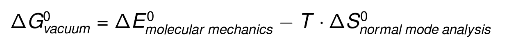
熵贡献可以通过对三种状态进行正态模式分析来发现,但实际上如果只需要比较相似熵的状态,例如两个配体结合同一蛋白质,则可以忽略熵贡献。 这样做的原因是正常模式分析计算的计算成本很高,并且往往具有很大的误差范围,从而在结果中引入了显着的不确定性。受体和配体的平均相互作用能通常是通过对从平衡分子动力学 (MD) 模拟收集的不相关快照集合进行计算来获得的。
- 介绍2
参考文献:Jawad, B., et al. (2021). "Key Interacting Residues between RBD of SARS-CoV-2 and ACE2 Receptor: Combination of Molecular Dynamics Simulation and Density Functional Calculation." J Chem Inf Model 61(9): 4425-4441.

介绍3:

- 计算注意事项
Section1:构建起始结构并运行模拟以获得平衡系统。
Section2:运行生产模拟并获得一组快照。
前两个部分就是跑MD,没什么可说的。有一点需要注意的是在生成拓扑的时候,有一个set default PBRadii mbondi2。设置这个的作用是为之后的GB计算设置正确的半径,比如GB模型使用igb=2或者igb=5的时候,参考半径为mbondi2。这里可以参考手册MMGBSA介绍部分关于igb的设置部分。
PS:如果最初在生成拓扑的时候没有设置这个也没问题,只要获得了动力学轨迹就行,可以在计算自由能的时候重新生成拓扑。————因为设置PBRadii mbondi2 这一项是用来隐式模型计算极性溶剂化自由能的,只是在GB计算部分要用到。而动力学轨迹是用来计算分子力学能的,所以这样做是没有问题的。
Section3:计算结合自由能并分析结果
-
Section 3.1 : Calculate the binding free energy of a protein-protein complex (Ras-Raf).
这个值得注意的是在运行$AMBERHOME/bin/MMPBSA.py -O -i mmpbsa.in -o FINAL_RESULTS_MMPBSA.dat -sp ras-raf_solvated.prmtop -cp ras-raf.prmtop -rp ras.prmtop -lp raf.prmtop -y *.mdcrd 的时候,-sp指定溶剂化复合物的拓扑的作用就是为了方便直接利用后面-y指定的动力学轨迹*.mdcrd。如果我们是跑完动力学后重新生成的受体配体复合物轨迹,就可以只使用-cp,-rp,-lp指定受体,配体,以及受配复合物的拓扑就行了,后面-y跟的也必须是受体配体复合物轨迹。
-
Section 3.2 : Calculate the binding free energy of a protein-ligand complex (Estrogen Receptor and Raloxifene).
这个和蛋白-蛋白计算差不多,没什么可讲的
-
Section 3.3 : Calculate the binding free energy of Ras-Raf and use Alanine Scanning to compare to the binding energy of a mutant Ras-Raf complex that has had a residue mutated to alanine and analyze the results.
丙氨酸扫描,Note that only one mutation can be performed during a single calculation.
最终获得的RESULT OF ALANINE SCANNING: (I21A MUTANT:) DELTA DELTA G binding = 2.1190 +/- 11.5368 为突变后的自由能-突变前的自由能,也就是DELTA DELTA G为正值说明突变后自由能大了,即该突变位点有利于结合。
-
Section 3.4 : Calculate the binding free energy of Ras-Raf in parallel using three processors.
例如使用mpirun -n 50 MMPBSA.mpi就行。需要注意的是使用的线程数必须要小于帧数,且最好能被帧数整除。
-
Section 3.5 : Calculate the entropy of the Estrogen Receptor and Raloxifene complex using Normal Mode Analysis (Nmode).
注意:All entropy results have units kcal/mol. (Temperature has already been multiplied in as 300. K)
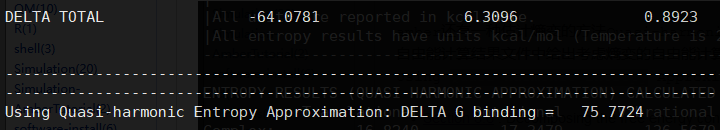
也就是如果不考虑熵变已经得到了结合自由能deltG,现在又得到了 T*熵变=deltS,根据刚开始介绍的公式,那么总结合自由能为deltG-delt。
PS:amber还有一种估计熵变的方法Quasi-harmonic Entropy Approximation,在mmpbsa.in文件中的&general中设置entropy=1,会在
自由能计算结果文件中给出熵变计算结果,即:
![]()
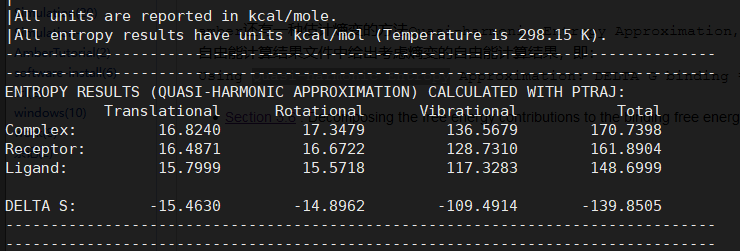
以及考虑熵变的自由能计算结果(内能-熵变),即:
![]()
-
Section 3.6 : Decomposing the free energy contributions to the binding free energy of Ras-Raf in a per-residue or pairwise per-residue basis.
能量分解,得到单个,或者成对残基对自由能的贡献。需要注意的是成对残基分解的时候,计算复杂度为n平方,PB模型非常之慢,最好不用
对结合自由能的理解可参考:https://blog.csdn.net/rogerzhanglijie/article/details/8126864
本文来自博客园,作者:计算之道,转载请注明原文链接:https://www.cnblogs.com/jszd/p/15574301.html

 浙公网安备 33010602011771号
浙公网安备 33010602011771号