GEO数据库转录组芯片数据处理与R分析:以GSE65682为例
找到感兴趣的GEO数据集后,如何从GEO网站上根据数据集编号下载呢?并且下载后怎么在R中对数据集进一步处理成后续分析所要的形式呢?以数据集(GSE65682)为例,为大家详细演示下如何使用R script 窗口的脚本进行下载和分析。
1.数据集获取
首先进入GEO网站官网(如下图所示),在检索位置输入数据集编号,点击箭头指向的位置进一步运行搜索。

搜索之后会弹出如下界面,重点检查物种类型(Homo sapiens)和数据集的类型,演示的数据集是芯片数据(Expression profiling by array)。

页面向下滚动,如图所示,展示了该数据集对应的注释文件及数据集内包含的样本。其中,注释文件GPL13667是我们重点关注的对象。
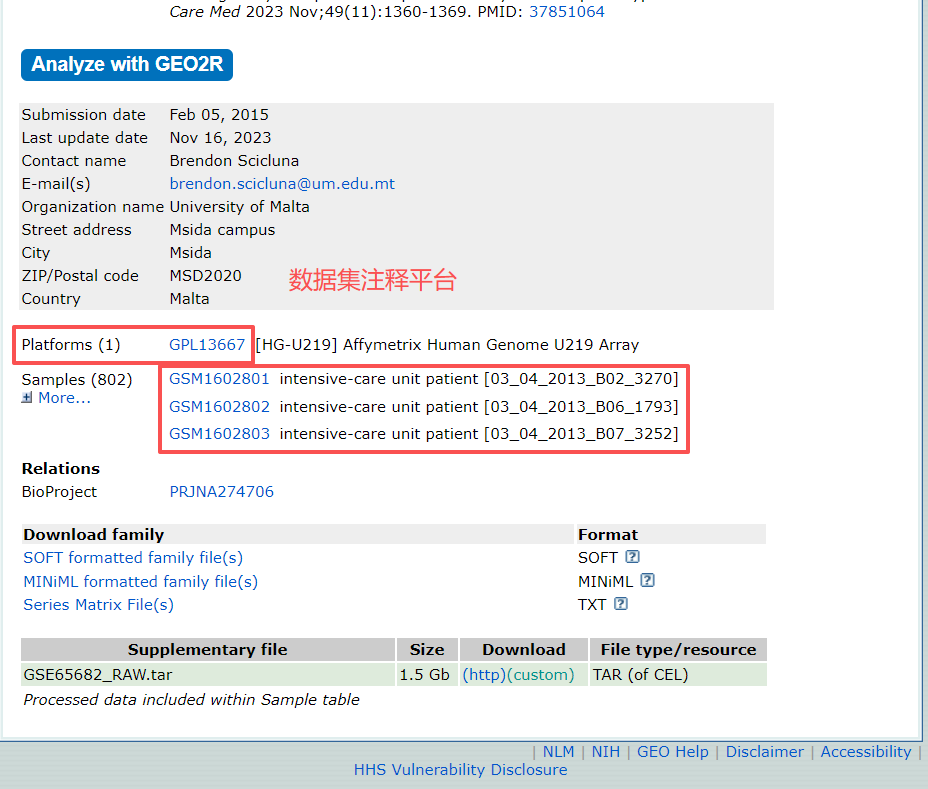
点击GPL13667可查看注释文件信息。将页面滚动至“Data table header descriptions”部分,快速了解注释文件所包含的信息。如图所示,该注释文件中包含了芯片的探针ID及其对应的基因符号(gene symbol),这些信息将在后续分析中被用到。
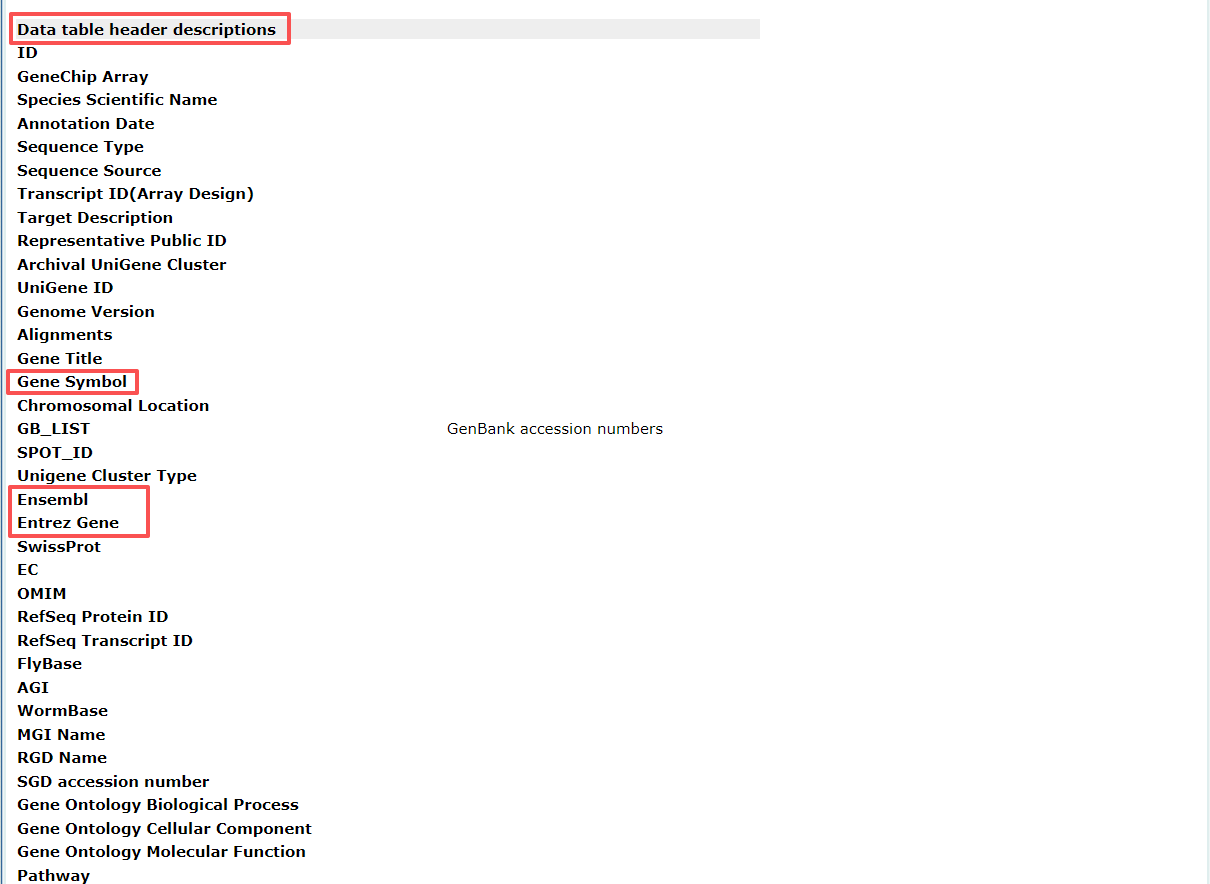
注意:GEO数据集如果只包含探针ID和对应的ENTREZID,则需要先将探针ID 转换为ENTREZID,再将ENTREZID转换为gene symbol。
2. 数据下载与处理
获取以上信息后即可直接进入R用代码自动下载数据
获取数据:
下载安装相关的R包;
library(BiocManager)
install("GEOquery")
加载所需R包;
library(GEOquery)
library(limma)
library(affy)
library(data.table)
library(dplyr)
链接GEO,在线下载数据集和注释文件(探针ID转symbol)
下载数据集
gset <- getGEO('GSE65682', destdir=".", # 下载到当前目录
AnnotGPL = TRUE, ## 下载注释文件
getGPL = TRUE, ## 获取平台信息
GSEMatrix = TRUE) ## 以GSEMatrix格式获取数据
获取表达矩阵数据
exp <- exprs(gset[[1]])
获取到的表达矩阵行名为芯片的ID,列名为样本ID。

获取样本的临床信息和注释文件
获取样本的临床信息
cli <- pData(gset[[1]]) ## 获取临床信息
获取平台注释信息(探针与基因的对应关系)
GPL <- fData(gset[[1]]) ## 获取平台信息
提取平台信息中的探针ID和基因符号列
gpl <- GPL[, c("ID", "Gene Symbol")]
清洗基因符号列:对于多个基因符号用"/// "分隔的情况,只取第一个基因符号
gpl$"Gene Symbol" <- data.frame(sapply(gpl$"Gene Symbol", function(x) unlist(strsplit(x, "/// "))[1]), stringsAsFactors = F)[, 1]
去除基因符号前后的空格
gpl$"Gene Symbol" <- trimws(gpl$"Gene Symbol")
将表达矩阵转换为数据框格式
exp <- as.data.frame(exp)
在表达矩阵中添加探针ID列
exp$ID <- rownames(exp)
将表达矩阵与平台注释信息按探针ID合并
exp_symbol <- merge(exp, gpl, by = "ID")
移除包含NA值的行
exp_symbol <- na.omit(exp_symbol)
检查基因符号的重复情况
table(duplicated(exp_symbol$"Gene Symbol"))
对重复的基因符号取平均值(去重)
exp_unique <- avereps(exp_symbol[, -c(1, ncol(exp_symbol))], ID = exp_symbol$"Gene Symbol")
移除基因符号为"---"的行(无效基因符号)
exp_unique <- exp_unique[row.names(exp_unique) != "---", ]
保存结果
write.csv(exp_unique,"GSE65682_exp_unique.csv")
提取临床信息中的样本ID、28天死亡事件和生存时间列
group_info <- as.data.frame(cli[, c(1, 52, 55)])
根据28天死亡事件信息创建分组变量,无事件为健康组,1为死亡,其他为存活
group_info <- group_info %>%
mutate(group = ifelse(mortality_event_28days:ch1 == "NA", "Healthy",
ifelse(mortality_event_28days:ch1 == "1", "Dead","Alive")))
重命名列使其更易读
group_info <- group_info %>% rename("sample" = "...1",
"status" = "mortality_event_28days:ch1",
"time" = "time_to_event_28days:ch1")
保存结果
write.csv(group_info,"GSE65682_group_info.csv",row.names = FALSE)
读取之前保存的表达矩阵数据
expr_data <- read.csv("GSE65682_exp_unique.csv")
读取分组信息
group_info <- read.csv("GSE65682_group_info.csv")
获取所有唯一的样本ID
sample_ids <- unique(group_info$sample)
将表达矩阵的行名设置为第一列(基因名)
rownames(expr_data) <- expr_data$X
提取表达矩阵中与分组信息匹配的样本列
expr_data_subset <- expr_data[, colnames(expr_data) %in% sample_ids]
从分组信息中筛选出非健康样本(败血症样本)
group_sepsis <- group_info %>%
filter(group != "Healthy")
获取疾病组样本的ID
sample_id <- unique(group_sepsis$sample)
提取疾病组样本的表达数据
exp_sepsis <- expr_data_subset[, colnames(expr_data_subset) %in% sample_id]
创建只包含样本和分组信息的简化数据框
group <- group_info[c("sample", "group")]
转置表达矩阵(样本为行,基因为列)
sample_exp <- t(expr_data_subset)
转换为数据框格式
sample_exp <- as.data.frame(sample_exp)
将行名(样本ID)转换为数据框的一列
sample_exp <- tibble::rownames_to_column(sample_exp, var = "sample")
将分组信息与表达数据按样本ID进行匹配
joined_df <- group %>% right_join(sample_exp, by = "sample")
保存结果
write.csv(joined_df,"GSE65682_exp_group.csv",row.names = F)
在得到处理后的表达矩阵以及临床信息后,就可以进行后续的很多个性化分析了,比如差异分析、生存分析,以及风险模型的构建等下游分析,因此这最初的第一步也是最重要的一步。
感谢大家的观看!以上内容涵盖了从GEO数据库下载转录组芯片数据到进行数据处理的完整流程。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号