从Hadder看蛋白质分子中的加氢算法
 本文主要介绍了开源加氢软件Hadder中用到的一些常规的补氢算法。在存储和优化蛋白质结构的过程中,人们更多的关注于蛋白质本身的骨架的变化,而单个原子的细微变化,对整体产生的性质是微乎其微的。但是我们在建立力场以及做能量最小化的过程中,需要用到氢原子。而氢原子的初始位置是至关重要的,如果加的位置太差,有可能导致体系能量过大,从而出现梯度爆炸的问题。
本文主要介绍了开源加氢软件Hadder中用到的一些常规的补氢算法。在存储和优化蛋白质结构的过程中,人们更多的关注于蛋白质本身的骨架的变化,而单个原子的细微变化,对整体产生的性质是微乎其微的。但是我们在建立力场以及做能量最小化的过程中,需要用到氢原子。而氢原子的初始位置是至关重要的,如果加的位置太差,有可能导致体系能量过大,从而出现梯度爆炸的问题。
技术背景
PDB(Protein Data Bank)是一种最常用于存储蛋白质结构的文件。而我们在研究蛋白质构象时,往往更多的是考虑其骨架,因此在很多pdb文件中直接去掉了氢原子。但是在我们构建蛋白质力场时,又需要用到这些氢原子。因此这个流程就变成了,在预测蛋白质构象时,不考虑氢原子,然后在力场构建的步骤去添加氢原子。由于氢原子的位置相对其连接的重原子来说,是相对比较固定的,而且最低能量位置也比较容易找到。因此常见的策略是,先在大致合理的位置补充上氢原子,再通过能量优化算法去优化氢原子的位置,使其处于一个更加合理的最终位置。而我们得到了这个氢原子的最终位置和重原子的位置之后,就可以对该蛋白质进行分子动力学的演化。本文主要介绍上述提到的,为蛋白质分子在大致合理的位置添加氢原子的算法。
效果预览
这里我们先看下加氢前后的效果,使用的工具是开源的轻量级加氢软件Hadder和分子动力学模拟常用的可视化软件VMD。在加氢之前,蛋白质的结构如下图所示:
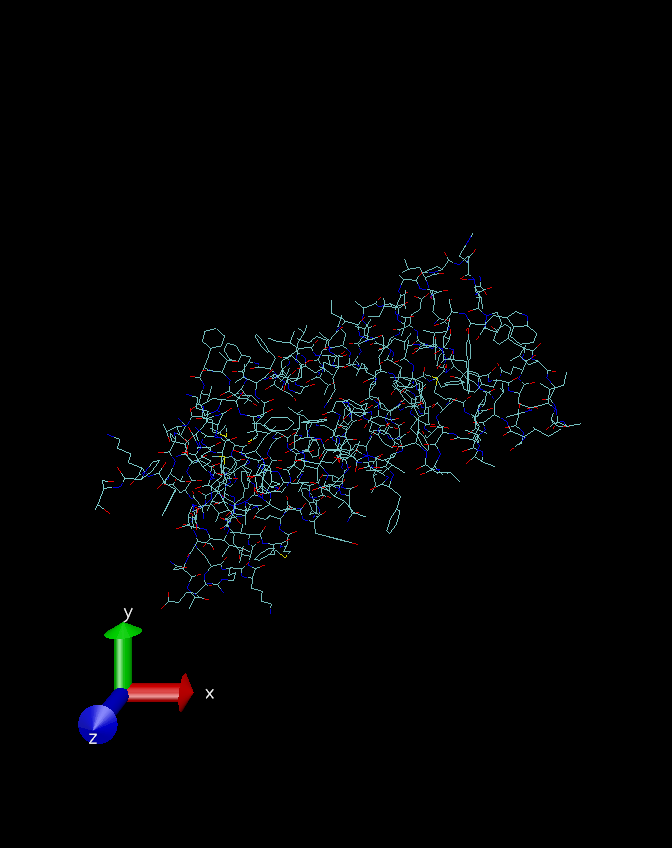
这是线条模型展示的结构,一般氢键用白色的线条来表示,可以看到上图中并没有白色的线条出现。其中有很多六边形的结构,其实就是苯环。再看一下加完氢原子之后的线条模型:
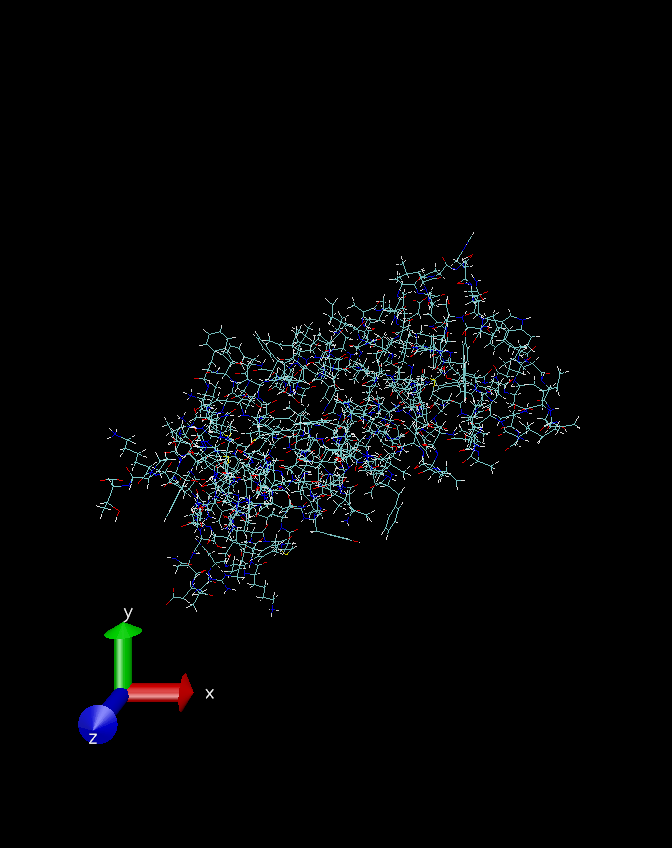
可以看到图中多出来了很多白色的线条,也就是补完氢原子之后生成的氢键了。关于氢原子所加的位置是否合理,可以参考其中苯环上加的氢原子和常见的甲基上添加的氢原子。
加氢算法
我们首先看一下GROMACS中所使用的加氢算法,大体上来说就是,将20种氨基酸中的氢原子所连接的重原子的拓扑结构枚举出来,然后针对这每一种的拓扑结构去执行不同的加氢方案。

那么,基于GROMACS的这个方案,我们对其进行了稍微的调整,也分成了以下6个不同的加氢方案来实施。需要说明的是,以下几个加氢方案的前缀命名是笔者随便取的,并不是什么规范性的要求,在Hadder中就是用了以下几种命名来作为算法索引。
1. c6--环结构补氢
不论是五元环还是六元环,其加氢的方式就是在最近邻三个点构成的平面的角平分线上,并且保障所加的氢原子与其所连接的重原子的距离为0.1nm。
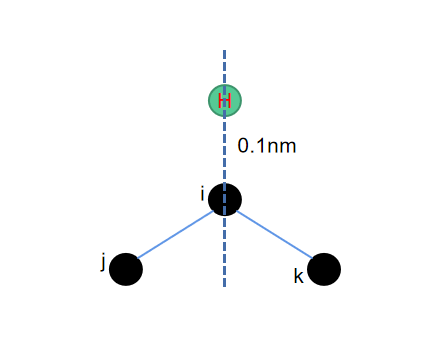
而这里寻找角平分线的算法也并不难,在python中只需要把\(\vec{ij}\)和\(\vec{ik}\)这两个矢量归一化之后,相加再取一个相反反向,再归一化一次,就可以得到需要添加氢原子的位置了。需要注意的是,这里长度单位使用的是埃而不是纳米,因此找到长度为1埃的氢键即可。
if type == 'c6':
left_arrow = crd[j] - crd[i]
left_arrow /= np.linalg.norm(left_arrow)
right_arrow = crd[k] - crd[i]
right_arrow /= np.linalg.norm(right_arrow)
h_arrow = -1 * (left_arrow + right_arrow)
h_arrow /= np.linalg.norm(h_arrow)
2. dihedral--二面角补氢
在上一个环结构中氢原子所连接的重原子,同时与其他另外两个原子相连接,而本章节中二面角形式的氢原子所连接的重原子,只与一个另外的重原子相连。同样的我们需要保障补充的氢原子跟这三个重原子处在同一个平面内。然后保障二面角的中心旋转对称性,就可以找到需要添加氢原子的位置。
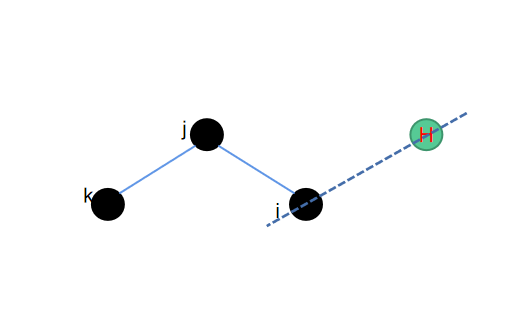
因为依然是在同一个平面内进行处理,因此也有比较简单的操作可以实现,相应的python代码如下:
if type == 'dihedral':
h_arrow = crd[j] - crd[k]
h_arrow /= np.linalg.norm(h_arrow)
这里使用的方法就是把向量\(\vec{jk}\)平移了一下,保持一样的角度即可。跟GROMACS中的方法略有点不同的是,在这里我们不追求一定达到109度的角度,更多的是使用系统本身自带的向量来进行操作。当然,这样实现的前提是,我们所得到的蛋白质的重原子结构,已经是一个相对比较合理的构象,否则按照本算法实施也会出现一些问题。
3. c2h4--乙烯结构补氢
同样的还是一个平面内的操作,类似于乙烯的结构,我们在得到i、j、k、l这4个位置的原子坐标之后就可以通过平移来得到需要补充的两个氢原子的位置。
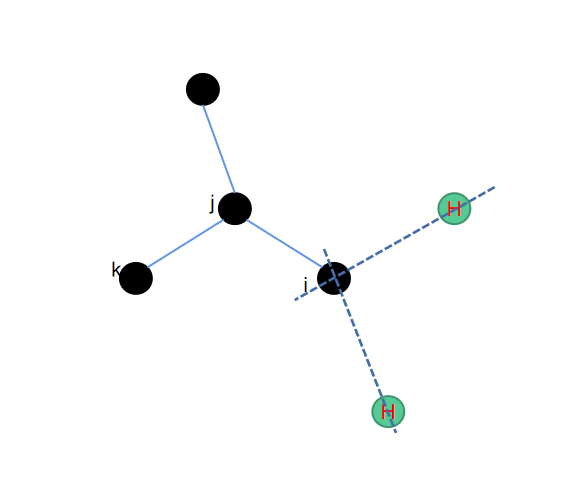
这里为了跟其他几种补氢的算法保持输入维度的一致,我们并没有直接将4个重原子的位置作为输入,而是取了其中的3个原子,再通过这3个原子的位置去推导氢原子的位置,相应python代码如下:
if type == 'c2h4':
h_arrow_1 = crd[j] - crd[k]
h1 = (h_arrow_1/np.linalg.norm(h_arrow_1) + crd[i])
middle_arrow = (crd[i] - crd[j])
middle_arrow /= np.linalg.norm(middle_arrow)
middle_arrow *= np.linalg.norm(h_arrow_1)
h_arrow_2 = -h_arrow_1 + middle_arrow
h2 = (h_arrow_2/np.linalg.norm(h_arrow_2) + crd[i])
在具体算法中,第一个氢的位置可以用跟上一个章节中二面角一样的算法推导出来,而第二个氢的位置,我们是假设这个乙烯结构中每一条边的长度都大致相等,这样根据等边三角形的矢量闭环关系,可以推导出来第二个氢原子的位置。
4. ch3--正四面体补三氢
一个sp3杂化的碳原子可以连接其他的4个原子,而甲基\(-CH3\)是很常见的一种基团,我们需要在这个碳上面补3个氢原子。那么此时除了碳原子的位置,和碳原子直接相连的一个原子的位置之外,我们还需要一个额外的原子,就是碳原子的次近邻原子,这样我们就有了三个原子可以去构造一个平面。
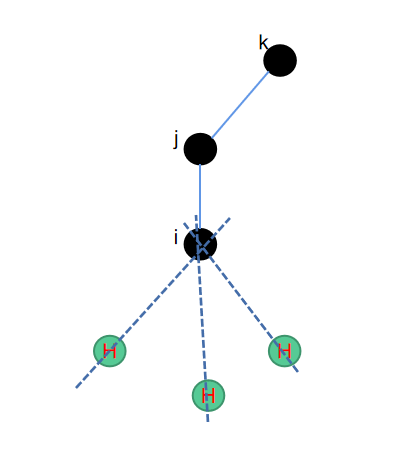
因为需要补氢的数量有3个,因此整体上算法会相对复杂一些。首先,补第一个氢原子位置时,可以参考二面角的补法,直接补上一个氢原子。补第二个和第三个氢原子时,需要用到一个平移旋转矩阵,其中又分为三个步骤:平移\(\vec{ij}\)到Z轴上->分别旋转第一个补的氢120度和240度->平移回原始的位置。相关python代码如下所示:
if type == 'ch3':
upper_arrow = crd[k] - crd[j]
upper_arrow /= np.linalg.norm(upper_arrow)
h1 = -upper_arrow + crd[i]
axes = crd[j] - crd[i]
rotate_matrix = rotate_by_axis(axes, 2 * np.pi / 3)
h2 = np.dot(rotate_matrix, h1-crd[i])
h2 /= np.linalg.norm(h2)
h2 += crd[i]
rotate_matrix = rotate_by_axis(axes, 4 * np.pi / 3)
h3 = np.dot(rotate_matrix, h1-crd[i])
h3 /= np.linalg.norm(h3)
h3 += crd[i]
这里还用到了一个旋转矩阵,其形式为:
相应的python代码实现为:
def rotate_by_axis(axis, theta):
"""Rotate an atom by a given axis with angle theta.
Args:
axis: The rotate axis.
theta: The rotate angle.
Returns:
The rotate matrix.
"""
vx, vy, vz = axis[0], axis[1], axis[2]
return np.array([[vx*vx*(1-np.cos(theta))+np.cos(theta),
vx*vy*(1-np.cos(theta))-vz*np.sin(theta),
vx*vz*(1-np.cos(theta))+vy*np.sin(theta)],
[vx*vy*(1-np.cos(theta))+vz*np.sin(theta),
vy*vy*(1-np.cos(theta))+np.cos(theta),
vy*vz*(1-np.cos(theta))-vx*np.sin(theta)],
[vx*vz*(1-np.cos(theta))-vy*np.sin(theta),
vy*vz*(1-np.cos(theta))+vx*np.sin(theta),
vz*vz*(1-np.cos(theta))+np.cos(theta)]])
类似的,这种算法对于初始构象有要求,如果是一个无序混乱的原子系统,则是没有办法通过这种算法来加氢的。
cc3--正四面体补一氢
还是sp3杂化的碳原子,但是此时该碳原子已经跟其他三个重原子成键,因此有一个多余的键可以跟氢原子结合生成氢键。由于sp3杂化的特殊性,形成的结构会是一个接近于正四面体的形状。此时同样为了保持接口一致,我们选取包含sp3碳原子在内的一共3个原子,来定位氢原子的位置。
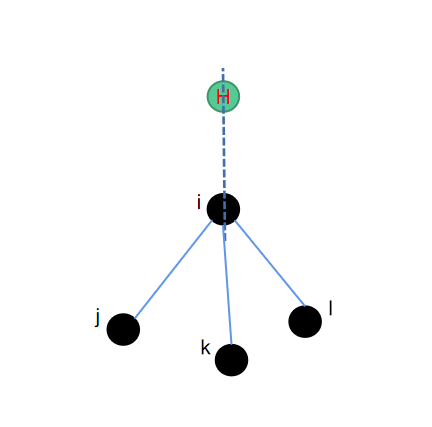
因为是一个正四面体,因此我们将\(\vec{ik}\)绕\(\vec{ij}\)逆时针旋转120度,即可得到氢原子的位置。其中用到的旋转矩阵可以参考上一个章节,对应的python代码实现如下:
if type == 'cc3':
h1 = crd[k]
upper_arrow = crd[j] - crd[i]
rotate_matrix = rotate_by_axis(upper_arrow, 2 * np.pi / 3)
h2 = np.dot(rotate_matrix, h1-crd[i])
h2 /= np.linalg.norm(h2)
c2h2--正四面体补二氢
从正四面体补三氢和补一氢的算法来看,我们还缺少一个补二氢的算法。跟补一氢的原理一样,也是找到三个重原子,然后对其中的一个键进行旋转。一次旋转120度,一次旋转240度,就可以得到待补的两个氢原子的位置。
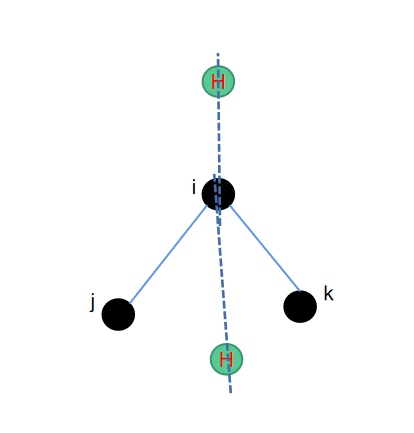
相应的python代码如下所示:
if type == 'c2h2':
right_arrow = crd[k] - crd[i]
rotate_matrix = rotate_by_axis(right_arrow, 2 * np.pi / 3)
h1 = np.dot(rotate_matrix, crd[j]-crd[i])
h2 = np.dot(rotate_matrix, h1)
h1 /= np.linalg.norm(h1)
h2 /= np.linalg.norm(h2)
当然,在实现的过程中,为了避免出现两个不同的旋转矩阵,这里只定义了一个旋转矩阵。旋转一次120度之后,对生成的新的氢键再绕相同的轴旋转120度,就可以得到第二个氢原子的位置。
Hadder的安装与使用
上述的这些补氢的算法,都已经实现在开源代码仓Hadder中,该代码都是基于python编写,开源依赖只有一个numpy。采用了开放式的开源协议Apache License 2.0。目前已经支持了pip的安装与管理,用户可以使用如下安装指令直接安装获取最新版本的hadder。
$ python3 -m pip install hadder --upgrade
因为只是为了给pdb补氢,因此软件中实现了pdb读取和写入的方法,而对外开放的API也较为简单,主要就是这样的一个补氢接口:
from hadder import AddHydrogen
AddHydrogen('input.pdb', 'output.pdb')
这样在python中定义好输入和输出的pdb文件路径(建议使用绝对路径),就可以获得补完氢原子的结果。如果运行成功,软件会有如下提示:
1 H-Adding task complete.
软件运行的效果在前面的章节中已经展示过了,这里再重复放两张图片:
分别对应的是加氢前与加氢后的结果。一个好的加氢位置,是至关重要的。如果一开始加氢的位置偏离较大,有可能导致体系能量异常的巨大,在计算梯度时会发生梯度爆炸的现象,也就无法正常的执行下一步的优化任务。
总结概要
本文主要介绍了开源加氢软件Hadder中用到的一些常规的补氢算法。在存储和优化蛋白质结构的过程中,人们更多的关注于蛋白质本身的骨架的变化,而单个原子的细微变化,对整体产生的性质是微乎其微的。但是我们在建立力场以及做能量最小化的过程中,需要用到氢原子。而氢原子的初始位置是至关重要的,如果加的位置太差,有可能导致体系能量过大,从而出现梯度爆炸的问题。
版权声明
本文首发链接为:https://www.cnblogs.com/dechinphy/p/hadder.html
作者ID:DechinPhy
更多原著文章请参考:https://www.cnblogs.com/dechinphy/
打赏专用链接:https://www.cnblogs.com/dechinphy/gallery/image/379634.html
腾讯云专栏同步:https://cloud.tencent.com/developer/column/91958
CSDN同步链接:https://blog.csdn.net/baidu_37157624?spm=1008.2028.3001.5343
51CTO同步链接:https://blog.51cto.com/u_15561675

 浙公网安备 33010602011771号
浙公网安备 33010602011771号