在甲基化ChAMP包中加入协变量
最近在分析甲基化数据,发现ChAMP包的输入端是beta(甲基化数据)和pheno(表型数据),找不到校正协变量(比如年龄、性别、批次等)的输入端。
如下代码所示:
champ.DMP(beta = myNorm,
pheno = myLoad$pd$Sample_Group,
compare.group = NULL,
adjPVal = 0.05,
adjust.method = "BH",
arraytype = "450K")
查了一下ChAMP包的代码,发现ChAMP包调用的是limma函数。
因此,这事就比较好解决了。
只需要在原代码的基础上修改model就行了。
下面介绍一下如何修改model。
1创建甲基化数据集和表型、协变量数据集
beta <- matrix(runif(60,min=0,max=1),10,6)
rownames(beta) <- c("cg18478105","cg09835024","cg14361672","cg01763666","cg12950382","cg02115394","cg25813447","cg07779434","cg13417420","cg12480843")
colnames(beta) <-paste("sample",1:6)
da=data.frame(pheno=c("1","1","1","2","2","2"),SEX=c("1","2","1","1","1","2"),AGE=c(54,23,58,43,68,36))
rownames(da) <- paste("sample",1:6)
甲基化数据如下所示:
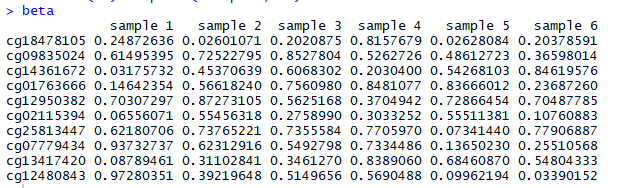
注意:beta数据是随机产生的,因此每个人生成的数据是不一样的。
表型(pheno)、协变量(SEX、AGE)的数据如下:
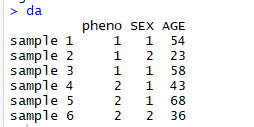
2 修改model、校正协变量
在这里我们假定要校正的协变量有SEX和AGE。
我在tian yuan创作的champ.DMP函数的基础上增加了cov1=da$cov1和cov2=da$cov2两个输入参数。
在champ.DMP函数里面修改了
design <- model.matrix( ~ 0+ factor(p)+cov1+cov2 )
colnames(design) <- c("control", "case","cov1","cov2")
contrast.matrix <- makeContrasts(contrasts=paste(colnames(design)[2:1],collapse="-"), levels = design)
最后修改完的函数如下所示:
champ.DMP1 <- function(beta = myNorm,
pheno = da$Sample_Group,
cov1=da$cov1,
cov2=da$cov2,
adjPVal = 0.05,
adjust.method = "BH",
compare.group = NULL,
arraytype = "EPIC")
{
message("[===========================]")
message("[<<<<< ChAMP.DMP START >>>>>]")
message("-----------------------------")
if(is.null(pheno) | length(unique(pheno))<=1)
{
stop("pheno parameter is invalid. Please check the input, pheno MUST contain at least two phenotypes.")
}else
{
message("<< Your pheno information contains following groups. >>")
sapply(unique(pheno),function(x) message("<",x,">:",sum(pheno==x)," samples."))
message("[The power of statistics analysis on groups contain very few samples may not strong.]")
}
if(is.null(compare.group))
{
message("You did not assign compare groups. The first two groups: <",unique(pheno)[1],"> and <",unique(pheno)[2],">, will be compared automatically.")
compare.group <- unique(pheno)[1:2]
}else if(sum(compare.group %in% unique(pheno))==2)
{
message("As you assigned, champ.DMP will compare ",compare.group[1]," and ",compare.group[2],".")
}else
{
message("Seems you did not assign correst compare groups. The first two groups: <",unique(pheno)[1],"> and <",unique(pheno)[2],">, will be compared automatically.")
compare.group <- unique(pheno)[1:2]
}
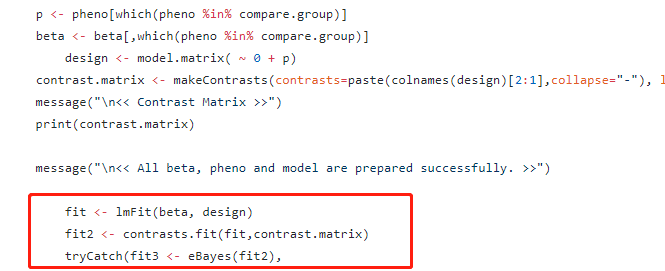
p <- pheno[which(pheno %in% compare.group)]
beta <- beta[,which(pheno %in% compare.group)]
design <- model.matrix( ~ 0+ factor(p)+cov1+cov2 )
colnames(design) <- c("control", "case","cov1","cov2")
contrast.matrix <- makeContrasts(contrasts=paste(colnames(design)[2:1],collapse="-"), levels = design)
message("\n<< Contrast Matrix >>")
print(contrast.matrix)
message("\n<< All beta, pheno and model are prepared successfully. >>")
fit <- lmFit(beta, design)
fit2 <- contrasts.fit(fit,contrast.matrix)
tryCatch(fit3 <- eBayes(fit2),
warning=function(w)
{
stop("limma failed, No sample variance.\n")
})
DMP <- topTable(fit3,coef=1,number=nrow(beta),adjust.method=adjust.method)
message("You have found ",sum(DMP$adj.P.Val <= adjPVal), " significant MVPs with a ",adjust.method," adjusted P-value below ", adjPVal,".")
message("\n<< Calculate DMP successfully. >>")
if(arraytype == "EPIC") data(probe.features.epic) else data(probe.features)
com.idx <- intersect(rownames(DMP),rownames(probe.features))
avg <- cbind(rowMeans(beta[com.idx,which(p==compare.group[1])]),rowMeans(beta[com.idx,which(p==compare.group[2])]))
avg <- cbind(avg,avg[,2]-avg[,1])
colnames(avg) <- c(paste(compare.group,"AVG",sep="_"),"deltaBeta")
DMP <- data.frame(DMP[com.idx,],avg,probe.features[com.idx,])
message("[<<<<<< ChAMP.DMP END >>>>>>]")
message("[===========================]")
message("[You may want to process DMP.GUI() or champ.GSEA() next.]\n")
return(DMP)
}
3、使用champ.DMP1分析甲基化数据
champ.DMP1 <- champ.DMP1(beta =beta,pheno = da$pheno, cov1=da$SEX,cov2=da$AGE, adjust.method = "BH", arraytype = "EPIC")
beta是第一步生成的甲基化数据,da数据框上的pheno、SEX、AGE也是在第一步生成的数据;
arraytype = "EPIC"表示甲基化数据类型为850K。如果是"450K"数据则将"EPIC"改为"450K";
4、修改技巧
上面举得例子是校正两个协变量,如果想校正三个协变量的话则做如下三个改动:
第一个改动:
champ.DMP1 <- function(beta = myNorm,
pheno = da$Sample_Group,
cov1=da$cov1,
cov2=da$cov2,
cov3=da$cov3,
adjPVal = 0.05,
adjust.method = "BH",
compare.group = NULL,
arraytype = "EPIC")
第二个改动:
design <- model.matrix( ~ 0+ factor(p)+cov1+cov2+cov3 )
colnames(design) <- c("control", "case","cov1","cov2","cov3")
contrast.matrix <- makeContrasts(contrasts=paste(colnames(design)[2:1],collapse="-"), levels = design)
第三个改动:
champ.DMP1 <- champ.DMP1(beta =beta,pheno = da$pheno, cov1=da$SEX,cov2=da$AGE, cov3=da$cov3,adjust.method = "BH", arraytype = "EPIC")
5、致谢
感谢原作者tian yuan(这个包很全能!);
感谢健明分享的甲基化分析入门练习:甲基化芯片的一般分析流程
建议各位刚入门甲基化的同学们可以看看健明在B站的视频,讲的很详细。

本文来自博客园,作者:橙子牛奶糖(陈文燕),转载请注明原文链接:https://www.cnblogs.com/chenwenyan/p/12920024.html

 浙公网安备 33010602011771号
浙公网安备 33010602011771号