关于train, evaluate 和 作图——dymean3
TMscore
从代码实现来看,这里的 TMscore 计算是通过调用外部的 TMscore 可执行程序完成的,输入的 PDB 文件包含了原子的三维坐标信息。那么具体答案可以分以下几个方面来分析:
1. 是否只对 CA 坐标进行计算
-
答案:否。
TMscore 通常会基于整个 PDB 文件中所有原子的坐标计算分子结构之间的相似性,而不是仅限于 CA 原子。虽然在某些情况下可以使用仅含 CA 原子的 PDB 文件,但这里的代码未对 PDB 文件进行显式简化(例如过滤掉非 CA 原子)。 -
PDB 文件的生成步骤:
mod_protein.to_pdb(mod_pdb) ref_protein.to_pdb(ref_pdb)这里没有显示提取特定的原子类型(如 CA 原子),意味着默认会写入所有原子。因此,
TMscore的计算会基于全原子信息。
2. TMscore 是对整条抗体链还是抗体-抗原进行计算
-
答案:整条抗体链。
代码中的tm_score函数明确指出,它将被调用时输入的是mod_cplx.antibody和ref_cplx.antibody。也就是说,TMscore的计算范围是 整个抗体链,并不包括抗原部分。在
tm_score函数中:results['TMscore'] = tm_score(mod_cplx.antibody, ref_cplx.antibody)mod_cplx.antibody和ref_cplx.antibody提供了抗体的坐标信息,而没有涉及抗原链。
3. TMscore 是否对抗体的某个区域进行计算
- 答案:否。
tm_score函数的输入是完整的mod_cplx.antibody和ref_cplx.antibody,它们代表完整的抗体结构,而不是特定区域(如 CDR 或框架区)。如果需要对特定区域计算 TMscore,需要手动提取这些区域的坐标并生成新的 PDB 文件,但代码中未执行这一步。
结论
- TMscore 默认计算的是整个抗体链的结构相似性。
- 它基于 PDB 文件中所有原子的坐标,而不仅限于 CA 坐标。
- 代码中的实现未对 TMscore 的计算范围进行细化(如局部区域或抗体-抗原相互作用)。
如果希望对抗体的某些特定区域(如 CDR)或者抗体-抗原界面计算 TMscore,需要对输入数据进一步处理。例如,仅生成感兴趣区域的 PDB 文件或局部坐标。
LDDT
从提供的代码来看,LDDT (Local Distance Difference Test) 的计算有以下几个关键点可以帮助回答您的问题:
1. 是否只对 Cα 坐标进行计算
-
答案:可选,但默认计算全原子信息。
-
代码中有一个关键参数
backbone_only:res_code = exec_bin(mod_pdb, ref_pdb, log, backbone_only)根据变量名
backbone_only的描述,如果设置为True,LDDT 的计算可能会被限制为只针对主链原子(N、Cα、C、O)。否则,默认情况可能是基于全原子计算。 -
LDDT 的实现(具体是通过
exec_bin函数调用的外部程序)未在代码中直接展现,但标准 LDDT 工具通常支持以下两种模式:- Backbone-only 模式:仅考虑主链原子(包括 Cα)。
- All-atom 模式:考虑所有原子的相对位置。
2. 计算范围是整条抗体链还是抗体-抗原
-
答案:整条抗体链。
-
merge_to_one_chain函数被调用后,将所有链拼接为单一的链:mod_protein = merge_to_one_chain(mod_protein) ref_protein = merge_to_one_chain(ref_protein)这说明,LDDT 的计算是基于整条抗体链的合并结果,不包含抗原。
-
如果需要计算抗体-抗原之间的 LDDT 分数,则需要在调用之前对输入数据进行筛选,将抗体和抗原的相关链分别提供。但当前代码显然是针对单一抗体链的整体计算。
3. 是否计算抗体的某个区域
-
答案:否,默认是整条抗体链。
-
从代码来看,输入是完整的
mod_protein和ref_protein,且没有特定区域的过滤逻辑。这意味着,LDDT 是基于整个抗体链进行计算的。 -
如果需要对抗体的某个区域(如 CDR 区域)计算 LDDT 分数,需要在
merge_to_one_chain或to_pdb调用之前,对抗体链提取感兴趣的部分(如 CDR 的残基范围)。
4. 外部程序的调用逻辑
- LDDT 的实际计算是通过调用外部程序实现的:
res_code = exec_bin(mod_pdb, ref_pdb, log, backbone_only)exec_bin具体的实现未在代码中提供,可能是一个对 LDDT 可执行文件(如lddt工具)的包装。- 具体 LDDT 的原理是通过比较模型和参考结构中每个原子的局部距离差异,得分是 0-1 之间的浮点数,越接近 1 表示模型和参考结构越一致。
5. 总结
- 是否只对 Cα 坐标计算:
- 默认是 全原子计算,但可以通过
backbone_only=True参数限制为主链原子(包含 Cα)。
- 默认是 全原子计算,但可以通过
- 计算范围:
- 当前代码计算的是 整条抗体链。
- 不涉及抗体-抗原或抗体局部区域。
- 如果需要计算抗体的某个区域(如 CDR)或抗体-抗原的局部 LDDT,需对输入数据进行额外处理。
如果您希望对抗体某个区域或者抗体-抗原的 LDDT 进行评估,可以通过提取感兴趣区域的残基坐标,然后重新生成 PDB 文件作为输入。
DockQ 函数分析
以下是对代码的详细分析,回答您的问题:
1. 是否只对 Cα 坐标进行计算
- 答案:否,DockQ 默认使用所有原子进行计算。
DockQ.py是一个独立的脚本,通常基于分子对接结果的接口原子来计算结构对齐度(包括界面原子和全原子)。- 代码中没有显式简化输入的 PDB 文件为仅含 Cα 的骨架结构,因此默认 DockQ 会使用 所有原子。
2. DockQ 的计算范围
- 答案:计算抗体-抗原界面区域的对接质量。
- 如果
cdrh3_only=False(默认设置),DockQ会对抗体-抗原整体界面进行评估,包括所有的界面原子。 - 如果
cdrh3_only=True,代码中明确提到只使用 CDRH3 区域(抗体的 CDRH3 和抗原的界面原子)进行 DockQ 计算:
这里将抗体的 CDRH3 和抗原链拼接为新的 PDB 文件用于计算,限制了计算范围。if cdrh3_only: mod_cdr, ref_cdr = mod_cplx.get_cdr(), ref_cplx.get_cdr() mod_peptides[H], ref_peptides[H] = mod_cdr, ref_cdr
- 如果
3. 总结分析
- 默认情况下,DockQ 计算抗体-抗原整体界面的对接质量。
- 如果指定
cdrh3_only=True,则计算范围限制在抗体的 CDRH3 区域 和抗原界面。 - 计算使用 所有原子,不局限于 Cα 原子。
DockQ vs TMscore vs LDDT 的区别
| 指标 | 是否只计算 Cα 坐标 | 计算范围 | 适用场景 |
|---|---|---|---|
| TMscore | 否,使用所有原子(但常用 Cα 坐标) | 整条抗体链 | 比较整条抗体链的全局结构相似性 |
| LDDT | 可选(通过 backbone_only=True) |
整条抗体链 | 评价整条抗体链局部结构的距离一致性 |
| DockQ | 否,使用所有原子 | 抗体-抗原界面(或抗体的特定区域,如 CDRH3) | 评价抗体-抗原的界面对接质量(结合几何和接触) |
使用建议
- TMscore:用于整条抗体链的全局结构比较(如模型和真实结构的全局一致性)。
- LDDT:用于评估整条抗体链的局部结构准确性(如主链一致性或原子间局部距离)。
- DockQ:用于评价抗体和抗原之间的对接质量,适合对抗体-抗原界面进行特定分析。
如果有其他需求(如针对特定区域计算),可以基于上述逻辑对 PDB 文件或输入数据进行筛选和处理。
作图
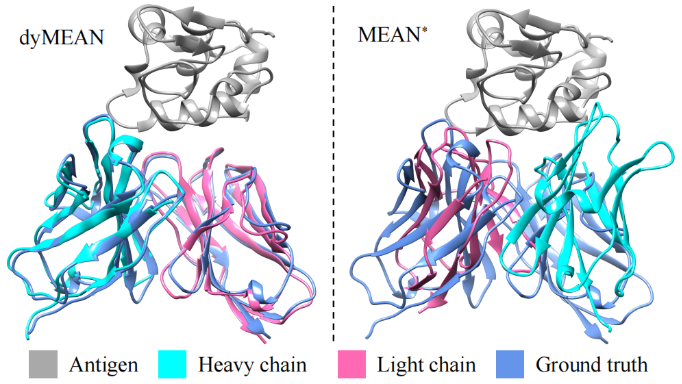
从图的特性来看,这种分子结构图通常是由 Cα 原子坐标 绘制的骨架表示(backbone representation)。以下是原因和分析:
1. 骨架图的常见特性
- 图中显示的分子结构清晰展示了蛋白质的 二级结构(如螺旋、β片层和环),这是典型的基于 Cα 原子绘制的骨架模型。
- 完整的原子坐标模型(包括侧链和其他原子)会包含更多的细节(如侧链和原子间的连接),而这里的图主要展示了蛋白质的主链。
2. Cα 坐标的使用
- Cα 是每个氨基酸主链上具有代表性的原子,通常用于表示蛋白质的整体形状和折叠。
- 绘制这种对比图时,Cα 坐标足以表现蛋白质的结构相似性,同时显著降低了计算复杂度。
3. DockQ 和其他指标的背景
- DockQ 指标常用于评价分子对接精度,可能基于所有原子计算,但绘图时更倾向于简化为主链模型(Cα 坐标)来更直观地对比。
- 图中高亮的抗体轻链、重链以及抗原的差异,主要依赖于二级结构对比,而这些信息也可以通过 Cα 坐标准确描述。
4. 常见软件实现
- 图像可能由 PyMOL、Chimera 或 VMD 等分子可视化工具生成,这些工具通常默认以骨架模型展示蛋白质结构。
- 如果要显示完整的原子信息,通常需要显式启用显示选项,但这会使图像显得复杂且冗余。
总结
图中结构图应该是基于 Cα 原子坐标 绘制的主链模型(backbone representation),未包含每个氨基酸的完整原子坐标。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号