100个GEO基因表达芯片或转录组数据处理26 GSE28623
100个GEO基因表达芯片或转录组数据处理
写在前边
虽然现在是高通量测序的时代,但是GEO、ArrayExpress等数据库储存并公开大量的基因表达芯片数据,还是会有大量的需求去处理芯片数据,并且建模或验证自己所研究基因的表达情况,芯片数据的处理也可能是大部分刚学生信的道友入门R语言数据处理的第一次实战,因此准备更新100个基因表达芯片或转录组高通量数据的处理。
数据信息检索
可以看到GSE28623是 芯片数据,因此可以使用GEOquery包下临床信息,然后从网页下载 原始的基因表达数据用 R 标准化处理

使用GEOquery包下载 临床 数据
BiocManager::install('ScienceAdvances/Canton')
Canton::using(using, tidyverse, GEOquery, magrittr, data.table, AnnoProbe, clusterProfiler, org.Hs.eg.db, org.Mm.eg.db,ggdendro,ComplexHeatmap)
注:using作用是一次性加载多个R包,不用写双引号,并且不在屏幕上打印包的加载信息
处理表型数据
这部分是很关键的,可以筛选一下分组表型信息,只保留自己需要的样本,作为后续分析的样本(根据自己的研究目的筛选符合要求的样本)
geo_accession <- "GSE28623"
eSet <- GEOquery::getGEO(geo_accession, AnnotGPL = F, getGPL = F)
pdata=Biobase::pData(eSet[[1]]) %>%
dplyr::mutate(
Sample = geo_accession,
Group = case_when(str_detect(`group:ch1`,"latently")~"LTBI",str_detect(`group:ch1`,"non-infected")~"HC",str_detect(`group:ch1`,"tuberculosis")~"TB",TRUE~NA),
Sex = str_to_title(`gender:ch1`)
) %>%
dplyr::select(Sample, Group,Sex, everything())
处理表达谱数据
读取原始数据
raw_dir <- "GSE28623_RAW"
x <- limma::read.maimages(
files = list.files(raw_dir, pattern = "^GSM", full.names = T),
source = "agilent",
green.only = TRUE,
other.columns = "gIsWellAboveBG"
)
使用 limma 标准化处理芯片数据
y <- limma::backgroundCorrect(x, method = "normexp") %>% limma::normalizeBetweenArrays(method = "quantile")
删除 Control 探针,没有基因名字的探针,重复的探针
Control <- y$genes$ControlType == 1L
NoSymbol <- is.na(y$genes$GeneName)
Isdup <- duplicated(y$genes$GeneName)
yfilt <- y[!Control & !NoSymbol & !Isdup, ]
获取表达量数据
fdata <- yfilt@.Data[[1]]
删除样本名中的路径字符串 GSE28623/GSM709520_251485039549_1_4.txt -> GSM709520
colnames(fdata) %<>% str_extract("GSM\\d*")
给数据框添加基因名作为行名
rownames(fdata) <- yfilt$genes$GeneName
数据质控
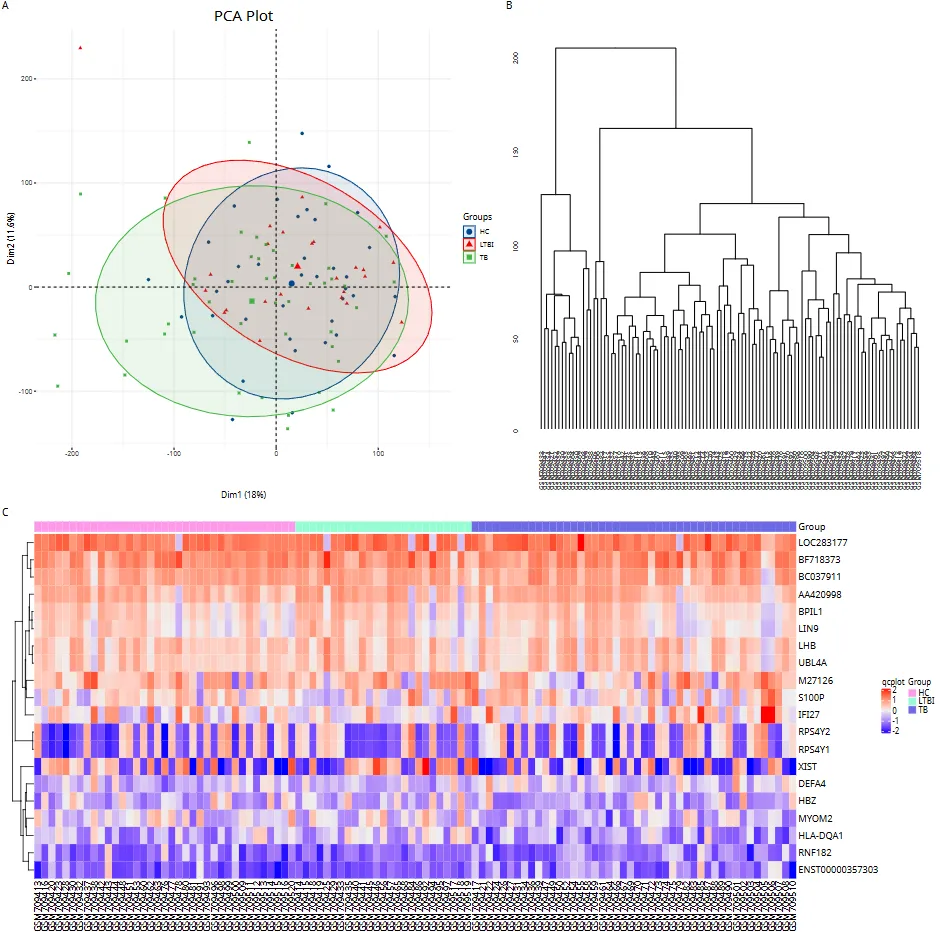
qcplot为自定义函数,作用是绘制用于质控判断的图,如PCA、top20基因热图、树状图,PCA图,可以三组区别区别不明显,可以考虑直接用作者处理好的数据虽然后负数值
qcplot(
data=fdata,
group=pdata$Group,
w=18,h=18
)
保存数据
common_samples <- base::intersect(colnames(fdata),pdata$Sample)
fdata=fdata[,common_samples]
fwrite(as.data.table(fdata,keep.rownames="Feature"), file = stringr::str_glue("{geo_accession}_fdata.csv.gz"))
pdata %<>% dplyr::filter(Sample %in% common_samples)
fwrite(pdata, file = stringr::str_glue("{geo_accession}_pdata.csv"))
Reference
https://www.jianshu.com/p/19ff109b6409

 浙公网安备 33010602011771号
浙公网安备 33010602011771号