100个GEO基因表达芯片或转录组数据处理025 GSE248467
写在前边
虽然现在是高通量测序的时代,但是GEO、ArrayExpress等数据库储存并公开大量的基因表达芯片数据,还是会有大量的需求去处理芯片数据,并且建模或验证自己所研究基因的表达情况,芯片数据的处理也可能是大部分刚学生信的道友入门R语言数据处理的第一次实战,因此准备更新100个基因表达芯片或转录组高通量数据的处理。
数据信息检索
可以看到GSE248467是 高通量测序数据,因此可以使用GEOquery包下临床信息,直接在页面下载 NCBI 生产的转录组表达矩阵 counts 和 tpm 两种进一步处理

使用GEOquery包下载 临床 数据
BiocManager::install('ScienceAdvances/Canton')
Canton::using(using, tidyverse, GEOquery, magrittr, data.table, AnnoProbe, clusterProfiler, org.Hs.eg.db, org.Mm.eg.db,ggdendro,ComplexHeatmap)
注:using作用是一次性加载多个R包,不用写双引号,并且不在屏幕上打印包的加载信息
处理表型数据
这部分是很关键的,可以筛选一下分组表型信息,只保留自己需要的样本,作为后续分析的样本(根据自己的研究目的筛选符合要求的样本)
geo_accession <- "GSE248467"
eSet <- GEOquery::getGEO(geo_accession, AnnotGPL = F, getGPL = F)
pdata=Biobase::pData(eSet[[1]]) %>%
dplyr::mutate(Sample = geo_accession,Group=stringr::str_split(pdata$title,"_",simplify=T) %>% .[,2]) %>%
dplyr::select(Sample,Group) %T>%
fwrite(file = stringr::str_glue("{geo_accession}_pdata.csv"))
处理表达谱数据
annot <- fread('Human.GRCh38.p13.annot.tsv.gz') %>%
dplyr::select(GeneID, Symbol )
counts=fread(glue::glue('{geo_accession}_norm_counts_TPM_GRCh38.p13_NCBI.tsv.gz')) %>%
merge(annot,.,by='GeneID') %>% dplyr::select(-GeneID) %>% unique_exprs %T>%
fwrite(file = stringr::str_glue("{geo_accession}_counts.csv.gz"))
tpm=fread(glue::glue('{geo_accession}_norm_counts_TPM_GRCh38.p13_NCBI.tsv.gz')) %>%
merge(annot,.,by='GeneID') %>% dplyr::select(-GeneID) %>% unique_exprs %T>%
fwrite(file = stringr::str_glue("{geo_accession}_tpm.csv.gz"))
数据质控
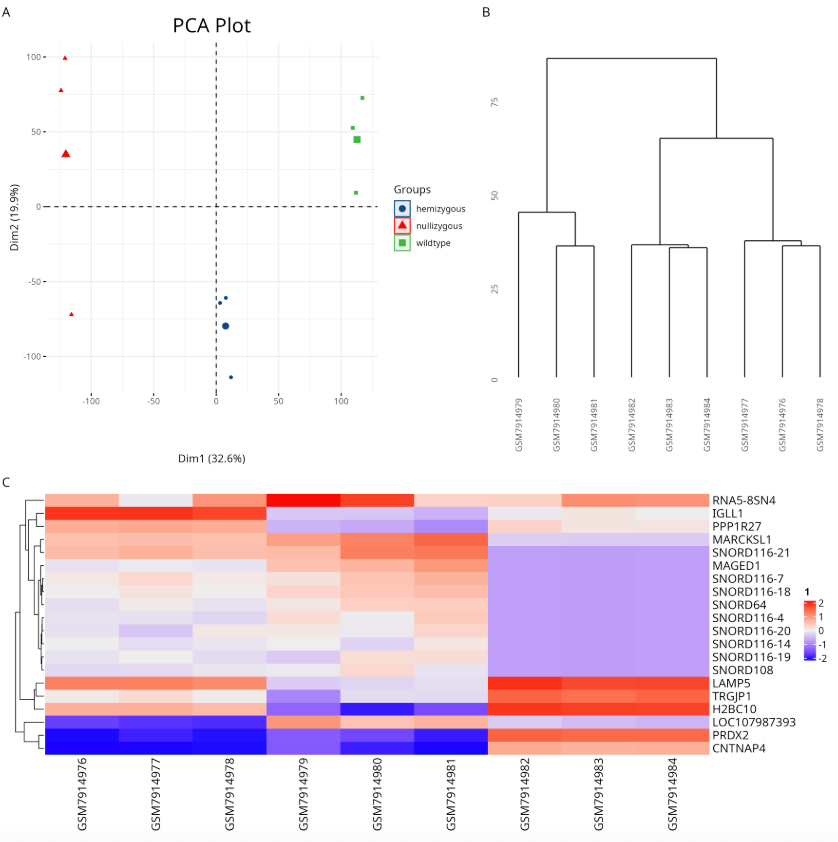
qcplot为自定义函数,作用是绘制用于质控判断的图,如PCA、top20基因热图、树状图,PCA图可以三组区别还是比较明显的
qcplot(
data=tpm %>% column_to_rownames('Symbol'),
pdata=pdata$Group,
w=12,h=12
)

 浙公网安备 33010602011771号
浙公网安备 33010602011771号