STRING Cytoscape 网络互作图
网络图(Network)看似复杂,其实构成非常简单,网络图是一种图解模型,形状如同网络,故称网络图,由节点(node)和连线(edge)两个因素组成的。其中 node 又分为 source node(源节点)和 target node(目标节点)两个因素组成的。这里的 node 就是我们的基因,edge 就是基因间的相互作用关系。任何网络图都不外乎这些构成成分。知道了网络图的构成之后,再做图分析就很简单了。
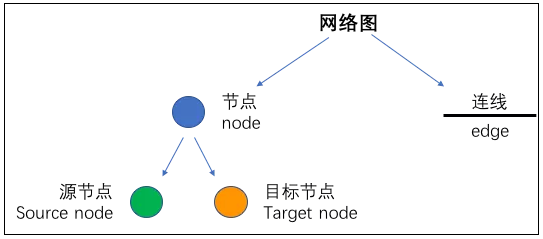
节点(node)
所谓的节点,就是我们要分析的基因。在一个网络图当中往往有数十个乃至上百个节点,那就意味着,我们需要分析的基因有数十个或者上百个。这些基因是怎么来的呢?这就跟我们的研究目的相关了,这些基因可能是我们筛选出来的差异表达的基因,可能是肿瘤患者中高频突变的一些基因,也可能是某一个 miRNA 的下游靶基因等等。
在进行网络图分析的时候,我们往往对基因的来源不做要求,只要是你认为是有意义的一群基因,就可以了。但是,在基因数量上,我们往往有一定的限制。因为,基因数少了,网络图中的 edge 太少,图做不出来,或者做出来太丑;而基因数多了,网络图太大,导致没办法导入软件中进行分析,耗时太久,同时背景噪声和混杂影响也会更多。因此,通常要分析的网络图基因数量在 50 - 300 个左右,这样的网络图比较适中,不会太大也不会太小。
连线(edge)
edge 就是基因之间的相互作用关系。比如两个基因 TP53 和 CXCL12 之间是否有相互作用关系呢?通过什么方法进行判断呢?这是一个比较困难的问题。好在,有一些非常好的数据库帮我们解决了这一问题,比如最著名的就是 STRING database。
STRING 数据库
STRING(https://string-db.org)是一个非常全面的蛋白互作网络数据库,里面存储了非常多物种和基因的相互作用关系。我们只要把基因名字提交上去,就能够判定他们之间时候有互作关系了。
STRING数据库是一个搜寻已知蛋白质之间和预测蛋白质之间相互作用的系统。这种相互作用既包括蛋白质之间直接的物理的相互作用,也包括蛋白质之间间接的功能的相关性,是目前最为全面、最为权威的蛋白相互作用数据库。
STRING数据库中包含有实验数据、从Pubmed摘要中文本挖掘的结果、综合其他数据库的数据,另外还有利用生物信息学的方法预测的结果,所应用的生物信息学的方法有:染色体临近、基因融合、系统进化谱、基于芯片数据的基因共表达等。
Cytoscape
Cytoscape是一套完整的网络图分析系统,它不仅仅是一个软件,还包括了一系列编程语言接口、app store 等诸多内容,是网络分析领域的龙头老大。Cytoscape 能够帮助我们实现基因互作的可视化网络图,并且通过其诸多分析插件帮我们找到这里面的关键基因。
研究思路
step1 从 基因列表 到 蛋白互作
step2 从 蛋白互作 到 互作网络
step3 从 互作网络 到 关键基因
具体步骤
step1 准备基因列表
这个基因列表的文件说白了就是一列基因,对于基因的数量最好是 50 - 300 个。
step2 打开 STRING 数据库
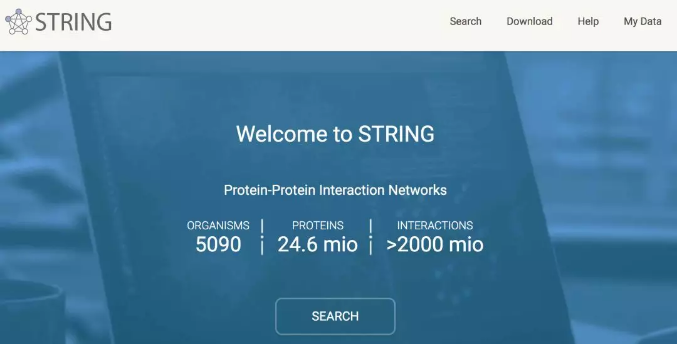
点击 SEARCH,然后就会跳转到让我们输入基因列表的页面,如下图所示,我们点击 "Multiple proteins",再依次输入我们的基因列表和物种名称,点击 SEARCH 即可。
然后 STRING 数据库会搜索我们提交的蛋白,点击 CONTINUE 即可。
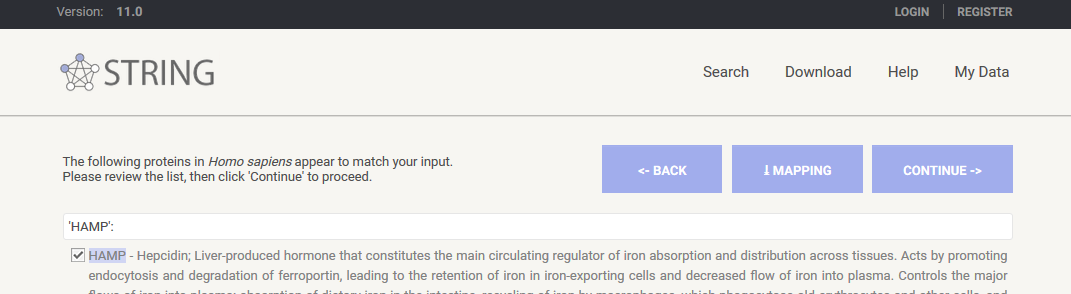
之后就会出现这些基因的互作网络图了。这个网络图中有很多彩色的点,这个颜色是随机分配的没有生物学意义,有的点中还有花花绿绿的蛋白质的三维结构,这个对我们来说也不是非常重要,重要的是蛋白之间的连线,这就是相互作用。
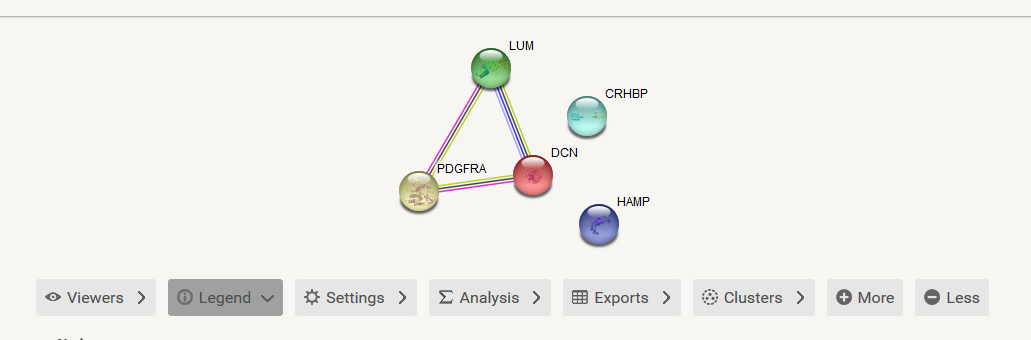
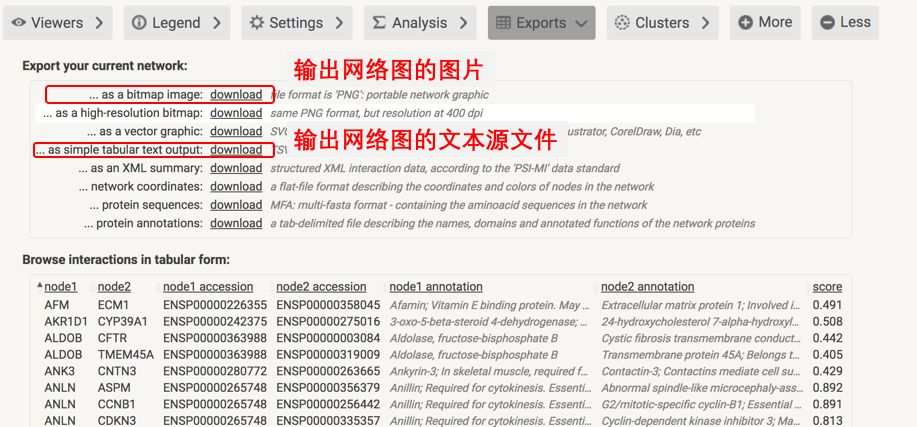
图的下面有很多的panel,这里面蕴含了很多功能,其中最主要的就是 Exports,从这里可以输出我们想要的图形和网络。
对于初级分析来说,网络图就可以了;当时如果是高级分析和美观的网络图,比如需要找到关键基因,需要发表质量的高级网络图,那就需要源文件了,源文件是一个tsv文件,通过它,可以制作各种各样的网络图。
step3 Cytoscape 美化网络图
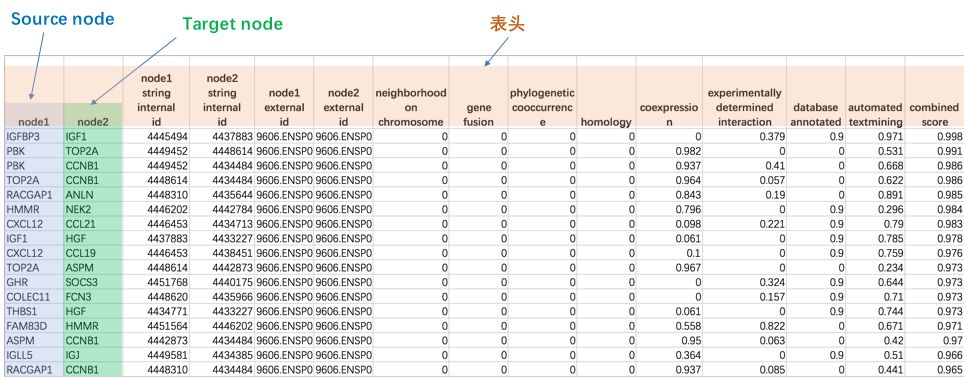
网络文件包括多种格式:TXT、SIF、GML等,这些都是Cytoscape能够识别的,其中最常用的就是TXT文本格式。这种格式是最简单的,其实就是从Excel中复制出来的表格,其格式如下:
(1)第一行,默认作为列名,所以不要有重复的名字
(2)从第二行开始就是节点之间的相互作用关系了
(3)数据至少包含两列,第一列是Source Node,第二列是Target Node
就拿我们从STRING生成的网络图源文件为例,我们生成的是一个名为string_interactions.tsv的文件,这是一个文本文件,用Excel把它打开之后是这样的
将该文件导入到Cytoscape中
File --> Import --> Network from File 即可导入文件
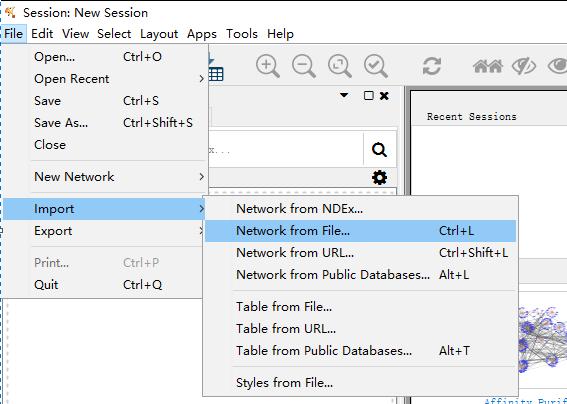
点击导入文件,找到要导入的网络文件,即string_interactions.tsv文件,导入之后是这样的。Cytoscape会自动识别最重要的两列:Source Node 和 Target Node,一般就是前面的两列。
如果自动识别的不对,可以自己指定。点击表头,会出现一个下拉菜单,然后自己选择指定即可。除了Source Node、Target Node 其他列数据的属性还包括Interaction Type、Edge Attribution、Source Attribution、Target Attribution等,同时对应不同颜色和图标标记。

指定好数据列之后,点击 OK 即可,数据导入到此为止,软件会自动生成一个网络图。
页面布局
Cytoscape是一个非常庞大的软件,其功能非常丰富,页面布局也很复杂,在我们导入网络图数据后,其会自动帮我们生成一个网络图,如下
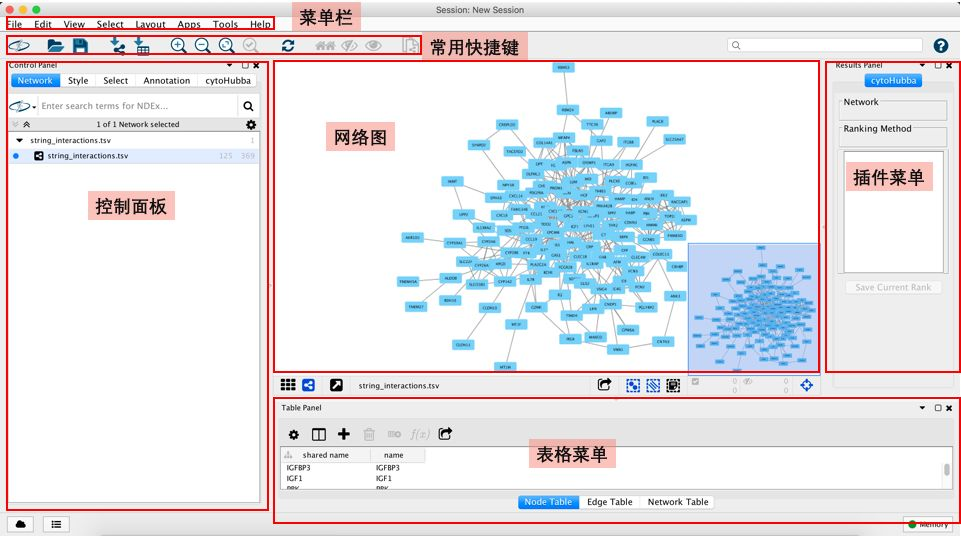
从上图中可以看出,页面相当复杂,而其中我们最常用的区域就是两个:控制面板和网络图区。
控制面板是我们用的最多的地方,这里面至少包括了三个子面板:
1)Network:网络图列表,这里以树形图的方式罗列了我们打开的所有网络图
2)Style:外观可视化控制面板,这个面板控制了我们的网络图的外观,所有的外观设计都在这个里面,这个面板也包括三个子面板:
-- Node:控制节点的外观,包括大小、颜色、形状等,使用频率很高
-- Edge:控制连线的外观,包括颜色、粗细等,使用频率很高
-- Network:控制网络图的外观,如背景色等,使用较少
3)Select:筛选,即从整个网络图中按照用户的要求去选定特定的Node或者Edge。
其实整个Cytoscape页面布局中最重要的就是控制面板,而通过控制面板中特定属性的设置,我们可以随心所欲的改变网络图的外观。

Cytoscape的应用商店
Cytoscape的菜单栏中有很多的功能栏,其中Apps就是很重要的一个,其提供了非常多的功能插件,使得Cytoscape的功能根据用户的需求无限延伸。

寻找关键基因
通过使用Cytoscape插件MCODE或者Cytohubba可以从网路图中找到关键基因
第一步 打开网络图
找到以 .cys 结尾的网络图文件,导入到Cytoscape中
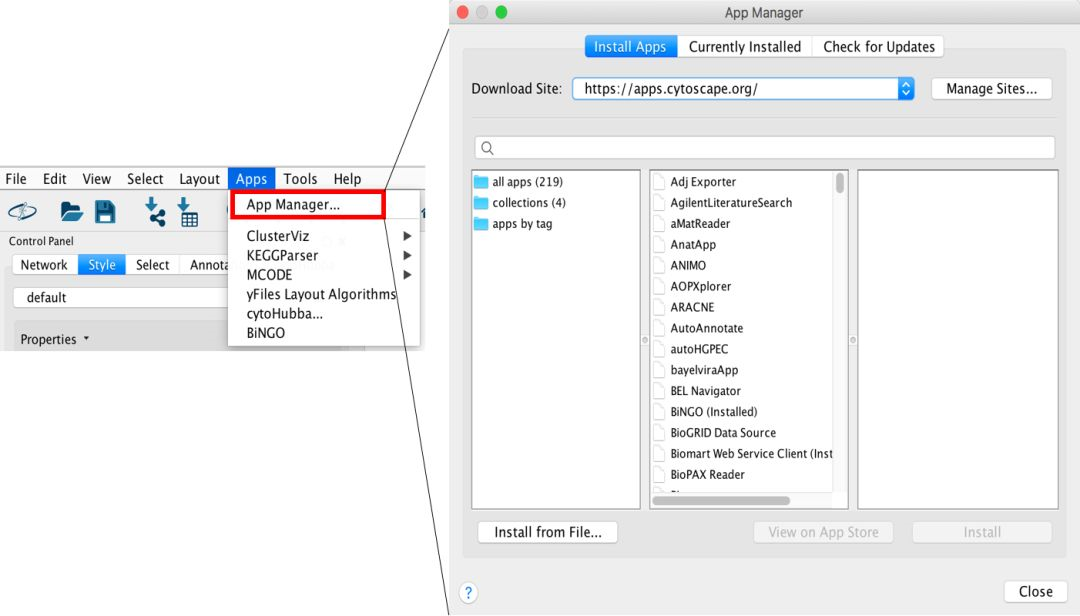
第二步 安装插件
点击 App Manager,然后经过一段时间的联网操作(注:有时候会经过很长时间连不上去,这时候需要耐心等待或者 VPN 操作一下),软件能够自动关联 App store,并且帮我们罗列出软件列表,如果我们要安装哪个软件,直接点击 Install 即可。
第三步 运行插件
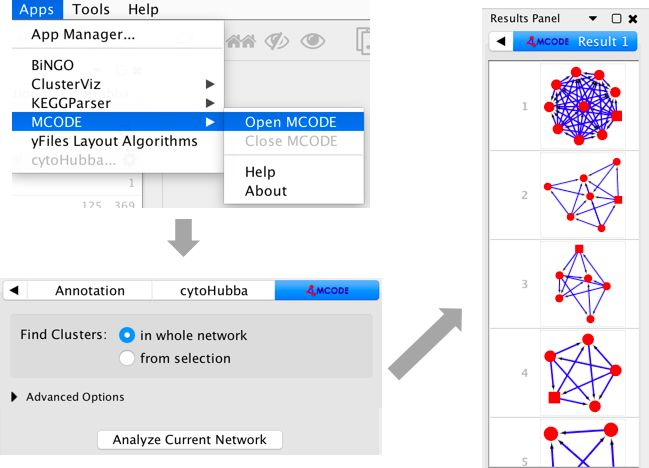
MCODE的使用
在 Apps 中点击 MCODE,然后会在控制面板中出现 Mcode这一面板,点击 Analyze Current Network 即可。

Cytohubba的使用
Cytohubba 的使用也比较简单,但是相对于 MCODE 来说,Cytohubba 提供了更多的算法来对基因的重要性或者说核心程度进行排序。使用 Cytohubba 的话,首先也是在 Apps 当中找到 Cytohubba,点击以后会在控制面板中出现Cytohubba 的子面板,然后按照我们下面的步骤操作,逐步点击即可:
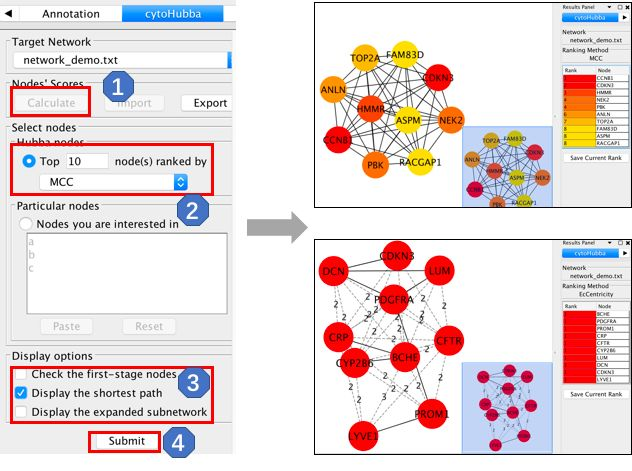
这里可以跟大家罗列一下Cytohubba所提供的核心基因筛选算法有哪些。算法虽多,但是这些算法的具体方法不需要我们大家掌握,只需要知道怎么用怎么选择就行了。


 浙公网安备 33010602011771号
浙公网安备 33010602011771号