
基于代谢基因共表达的一种新的更具信息量的乳腺癌亚型方案

https://www.sciencedirect.com/science/article/pii/S2352304225000789?via%3Dihub
1.研究背景和理论基础
- 现有分类方法的局限性
当前乳腺癌分类(如PAM50亚型)依赖少数标记基因或蛋白的表达,无法充分捕捉样本间共同的生物学特征,导致分类结果在临床应用中受限。 - 新分类方法的理论依据
- 代谢重编程是癌症的核心特征 :癌细胞通过改变代谢途径(如糖酵解、脂质合成)适应肿瘤微环境压力(如缺氧、营养缺乏),这是癌症的标志性特征之一。
- 代谢驱动癌细胞行为 :所有癌细胞的异常行为(增殖、侵袭、免疫逃逸)均与代谢重编程直接相关。
- 基因组规模代谢基因的稳定性 :基于代谢基因的表达进行分类,可减少随机噪声干扰,提高分类结果的稳定性。
- 新分类的优势
- 相比PAM50亚型,新分类(四类)在以下方面更具区分度:
- 代谢特征 :每类具有独特的代谢通路活性。
- 临床特征 :与患者预后、治疗反应更相关。
- 多组学特征 :整合基因组、表观组、蛋白组数据,揭示肿瘤微环境和免疫浸润差异。
- 相比PAM50亚型,新分类(四类)在以下方面更具区分度:
2. 研究方法与结果
- 数据来源与聚类方法
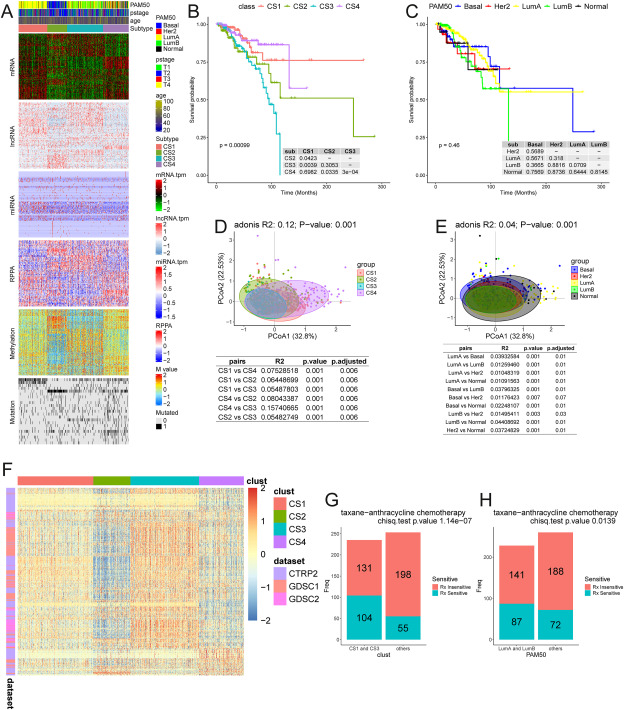
We classified the BC samples collected from TCGA databases using the clustering function of the R package MOVICS, consisting of ten clustering methods: iClusterBayes, SNF, PINSPlus, NEMO, COCA, LRAcluster, ConsensusClustering, IntNMF, CIMLR, and MoCluster, based on co-expressions of metabolic genes (Supplementary Methods; Table S1) plus additional omics data such as genomic, proteomic, and methylomic data. This led to four distinct classes: CS1, CS2, CS3, and CS4 (Fig. 1A)- 数据 :使用TCGA数据库的乳腺癌多组学数据(基因表达、基因组变异、甲基化、蛋白组)。
- 聚类工具 :R包MOVICS,集成10种机器学习方法(如iClusterBayes、SNF、ConsensusClustering等,综合多组学数据进行聚类。
- 关键步骤 :
- 迭代筛选特征基因:最终每类选定100个特异性上调的代谢基因。
- 使用“The nearest template prediction algorithm”确保分类稳定性(P < 0.001)。
- 通过轮廓系数(Silhouette Score)验证类内高相似性与类间显著差异。
- 分类结果与验证
- 四类分型(CS1-CS4) :
- 生存分析 :新分类的四类间生存时间差异显著,优于PAM50亚型(图1B, C)。
- 临床特征 :不同类与肿瘤分期、分级等临床指标关联(图S2A)。
- 与PAM50亚型的对应关系 :
- CS1:91%对应Luminal A(LumA)。
- CS2:74%对应Basal-like(三阴性乳腺癌)。
- CS3:混合LumA(53%)和Luminal B(LumB,42%)。
- CS4:分布于所有PAM50亚型,可能代表代谢异质性更高的群体。
- 稳定性验证 :
- 新分类与肿瘤分期无关(图S2B, C),表明代谢特征是肿瘤的内在属性,不随进展显著变化。
- 四类分型(CS1-CS4) :
- 生物学意义
- 代谢特征的主导性 :每类的100个标记基因反映其独特的代谢重编程模式(如糖酵解、氧化磷酸化、谷氨酰胺代谢)。
- 治疗意义 :不同类可能对化疗、免疫治疗(如免疫检查点抑制剂)的敏感性不同,为个性化治疗提供依据。
- 核心结论
- 代谢驱动分类 :基于代谢基因的分类更贴近肿瘤生物学本质,克服了传统分类的片面性。
- 临床实用性 :新分类在预后评估、治疗选择(如免疫治疗)中可能更具指导价值。
- 稳定性与普适性 :代谢特征在肿瘤不同阶段保持稳定,适用于动态监测和早期诊断。
- 技术术语解释
- 代谢重编程 :癌细胞通过改变代谢通路(如增强糖酵解)满足快速增殖的能量和生物合成需求。
- 多组学整合 :联合基因组、转录组、蛋白组等数据,全面解析肿瘤异质性。
- 机器学习聚类 :通过算法(如SNF、ConsensusClustering)识别数据中的潜在分组模式。
![]()
3.代谢通路差异与基因突变分析
- 代谢通路特征(图S3)
- 差异表达基因筛选 :以|log2(FC)|≥1且Padj<0.05为标准,识别各亚型特异性差异表达的代谢酶基因。
- 通路富集分析 :
- CS1 :中长链脂肪酸、花生四烯酸、丁酸代谢活跃,可能与激素依赖性生长相关(符合Luminal亚型特征)。
- CS2 :硫化合物、一碳代谢、糖蛋白合成、氨基酸代谢增强,可能支持快速增殖和侵袭(对应Basal-like亚型的高侵袭性)。
- CS3 :胰岛素代谢、氧化磷酸化、核苷酸从头合成活跃,提示能量代谢重塑和DNA合成需求(可能与Luminal B的高增殖性相关)。
- CS4 :免疫活性高,前列腺素、NAD+、色氨酸代谢显著,可能与炎症微环境及免疫逃逸相关。
- 与PAM50对比 (图S3F, G):新分类的代谢特征更清晰,而传统亚型的代谢差异被稀释。
- 基因突变特征(图S4)
- 突变率与肿瘤突变负荷(TMB) :
- CS2 突变率最高(TMB最高),可能因DNA修复缺陷导致基因组不稳定性。
- CS1 突变率最低,基因组较稳定(与Luminal A的低突变特征一致)。
- 突变基因的通路富集 :
- CS1 :细胞周期调控、DNA损伤检查点(提示增殖相关突变)。
- CS2 :神经发育与神经发生(可能与Basal-like亚型的基底上皮起源相关)。
- CS4 :表观遗传调控、转录调控(可能影响免疫微环境)。
- 结论 :突变基因的功能通路(而非单个基因)决定亚型表型,新分类的突变特征更显著(图S4G, H)。
- 突变率与肿瘤突变负荷(TMB) :
4. 免疫微环境与免疫治疗反应
- 免疫浸润与免疫治疗预测
- 免疫评分与功能障碍评分 :
- CS4 :免疫评分最高(高免疫浸润),但功能障碍评分也最高(免疫抑制微环境),可能对免疫检查点抑制剂(如PD-1/PD-L1抗体)反应差。
- CS2 :TMB高且功能障碍评分低(免疫逃逸少),可能对免疫治疗敏感。
- 与PAM50对比 (图1D, E):新分类在免疫特征上分层更清晰,传统亚型的免疫异质性较高。
- 免疫评分与功能障碍评分 :
5. 化疗敏感性验证
- 药物敏感性分析(GDSC/CTRP数据库)
- IC50预测 :各亚型对化疗药物(如紫杉烷-蒽环类)的敏感性差异显著。
- 临床验证(GSE25066队列) :
- CS1和CS3 (Luminal A/B为主)对紫杉烷-蒽环类化疗敏感性更高(P=1.14e-07)。
- 传统PAM50亚型 的敏感性差异较小(P=0.01385),表明新分类在预测化疗反应上更精准。
- 核心结论与意义
- 代谢通路主导分型 :
- 每个亚型的代谢特征与其生物学行为(增殖、侵袭、免疫逃逸)直接相关,为靶向代谢治疗提供依据。
- 基因组与免疫特征 :
- CS2 (高TMB+低免疫抑制)可能受益于免疫治疗;CS4 (高免疫抑制)需谨慎使用免疫检查点抑制剂。
- 临床应用价值 :
- 新分类在预测预后、化疗敏感性和免疫治疗反应上优于传统分型,有助于实现精准医疗。
- 代谢通路主导分型 :
- 技术术语解释
- TIDE评分 :预测肿瘤免疫逃逸潜力的指标,综合考虑免疫抑制和排斥机制。
- IC50 :药物抑制50%细胞活性的浓度,值越低表示敏感性越高。
- χ2检验 :用于比较分类变量(如化疗敏感性)的统计显著性。
- 研究亮点
- 多维度整合分析 :结合代谢、基因组、免疫和药物敏感性数据,全面解析肿瘤异质性。
- 临床可操作性 :为不同亚型提供明确的治疗策略(如CS2优先免疫治疗,CS1/CS3优先化疗)。
- 稳定性验证 :代谢特征在肿瘤不同阶段保持稳定,适用于动态监测。
这项研究通过代谢重编程重新定义乳腺癌亚型,为个性化治疗提供了更可靠的生物学基础,可能推动代谢抑制剂、免疫治疗和化疗方案的优化组合。
Materials and Methods
Study cohorts
该研究是基于来自TCGA的1,226个BC样本和113个癌旁组织的转录组数据。多组学数据类型,即转录组、基因组、甲基组、蛋白质组和临床数据,均来自TCGA官方网站。转录组数据也从GEO数据库中收集,包括两个BC数据集进行验证。GSE202203有3207个样本,METABRIC有2509个样本,GSE20685有327个样本,GSE25066有508个样本。使用R软件包GEOquery下载了这些GEO样本的转录组和临床数据。所有人类酶基因均来自HumanCyc(见补充表1)。转录组数据进行了log2(tpm+1)转化。基因组数据包括单核苷酸突变和拷贝数变异。
Classification method
对TCGA内的BC样本利用R包"MOVICS"基于转录组数据(tpm),基因组,蛋白组和甲基化数据,其中转录组数据包括mRNA(经log2转换),IncRNA和miRNA;基因组突变数据覆盖每个基因组的所有单点突变,蛋白质数据是反相蛋白阵列(Reverse Phase Protein Arrays).随后,我们使用MOVICS包中"getElite"函数来筛选特征基因,将参数“mad"设置为1500来筛选变异最大的前1500个基因,参数"COX"沿每个数据维度来筛选预后相关基因(P值小于0.05),参数“freq"为10%,以筛选最常见突变的前10%的基因。在“getMOIC”函数中选择10种聚类算法,在“方法列表”下选择以下方法:聚类、SNF、PINSPlus、NEMO、COCA、LRA聚类、共识聚类、IntNMF、CIMLR、移动聚类分析。对于其他参数,将使用MOVICS软件包提供的默认值。随后利用"getConsensusMOIC"函数对不同算法的结果进行整合以最大化这十种分类结果的一致性。然后,我们使用MOVICS包中的“getClustNum”函数来确定BC样本的集群数量,先验知识也被应用,从而产生了四类。
Differentially expressed genes
使用R包DEseq2对每个类的成员与对照组进行差异表达分析,以识别类特异性的差异表达基因。具有绝对log2(FC)值>为1.0和P.调整值<为0.05的基因被认为是差异表达的。
Pathway enrichment analyses
差异表达的基因需要对从KEGG11数据库(https://www.genome.jp/kegg/)和GSEA MSigDB网站12(http://gsea-msigdb.org/gsea)中检索到的代谢通路进行通路富集分析。差异基因采用基于代谢基因集的R聚类分析软件包进行GSEA分析。
Genomic mutation analysis
一个癌症类别的随机突变率定义为单点突变的总数除以相关基因组的总长度。如果一个BC基因组中的一个基因的突变数至少是随机突变率的三倍,则认为该基因被选择进行突变。R包“maftools”用于识别每个基因组中的前10个突变基因。
Target gene of miRNA
我们从mirDIP数据库中发现了与mirnas相对应的靶基因
Prediction of immune cell infiltration in cancer tissues肿瘤组织中免疫细胞浸润的预测
R包CIBERSORT用于估计以下特定癌症组织中的免疫细胞的relative abundance。包括有B naive cells,B memory cells,plasma cells(浆细胞),CD8 T细胞,CD4 T4 cells naive,CD4 memory resting T cells, CD4 memory activated T cells, T follicular helper cells, T regulatory cells (Tregs), gamma delta T cells, resting NK cells, activated NK cells, monocytes, macrophages M0, macrophages M1, macrophages M2, resting dendritic cells, activated dendritic cells, resting mast cells, activated mast cells, eosinophils, neutrophils based on the gene-expression data of BC tissues in the TCGA databases.
Prediction of immunotherapy response
特定癌症组织的TIDE评分通过TIDE网站获得,其中TIDE评分越高,说明癌症肿瘤具有较强的免疫逃逸能力,这与较差的免疫检查点抑制治疗反应有关
Assessment of similarity and difference between sample groups
采用基于欧氏距离的R包pairwiseAdonis进行多变量非参数检验分析。
Drug sensitivity prediction
我们根据TCGA和GEO样本的转录组数据预测药物敏感性,使用R包oncopredict和GDSC和CTRP数据库中的药物,计算每个样本的IC50得分。IC50得分越低,癌症组织对药物就越敏感。
Statistical analysis and mapping
所有计算均使用R(版本4.3.2)进行统计分析。两组与正态分布变量的比较采用非配对学生t检验,非正态分布变量采用Wilcoxon检验。总生存期(OS)分析采用生存期软件包进行,采用log - rank检验来确定生存期差异。p.<值为0.05,表示差异有统计学意义。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号