

swga 设计引物
该软件的使用非常方便,一般使用默认参数就可以出不错的结果。主要分为以下几步:
- 起始:在之前设置的虚拟环境中创建一个项目文件夹。以后我将把这个项目文件夹称为 SWGA_FOLDER(但您可以而且应该为您的项目命名一个合适的名称!)
$ mkdir SWGA_FOLDER
$ cd SWGA_FOLDER
$ swga init2. 计数:计算前景和背景基因组中的 kmers。默认的 kmer 大小范围是 5-12bp。您可以使用 --min_size 或 --max_size 参数更改它。这一步将需要一段时间,具体取决于您的背景基因组有多大,但请耐心等待 - 如果您重新运行此步骤,它会记住您已经计算过的大小。此外,它只保留与前景基因组至少结合几次而与背景基因组结合次数不多的 kmers。这可以防止我们花时间存储我们永远不会使用的 kmers。您可以在“parameters.cfg”文件中或作为命令选项更改这些截止值。
$ swga count3. 过滤:根据各种标准选择引物,例如最小前景结合频率、最大背景结合频率以及最小和最大熔解温度。所有这些标准都可以通过查看 SWGA_FOLDER 中 parameters.cfg 文件的 [filter] 部分找到,或者通过在 filter 命令后键入 --help 来找到。与所有命令一样,如果您未指定任何内容,它将使用“parameters.cfg”中的选项。
$ swga filter4. 查找集:现在我们实际上将使用我们在 swga 过滤器中识别的引物来查找兼容的引物集。对于非平凡数量的引物,可能的集合呈指数级增长,因此此命令需要很长时间。幸运的是,您不必等待它完成就可以开始使用它的结果。我们建议使用带有屏幕或 tmux 的终端,这样您就可以让它在后台运行。
$ swga find_sets5. 导出:您可以使用导出命令导出有关引物和集合的信息。例如,要获得排名最高的前 50 个集合:
$ swga export sets --order_by score --limit 50默认情况下,export 会将数据打印到屏幕上。要将其保存到文件中:
$ swga export sets --order_by score --limit 50 > best_sets.txt如果您想按 ID 获取一个集合(例如,swga 摘要所示的最佳集合),请改用 --id。更多示例:
# Top 100 primers with highest foreground/background binding ratios:
$ swga export primers --order_by ratio --descending --limit 100
# Get sets by ids:
$ swga export sets --id 5, 10, 15
# Get top 10 sets ordered by most even foreground binding site distribution:
$ swga export sets --order_by fg_gini_index --limit 10导出结果如图所示
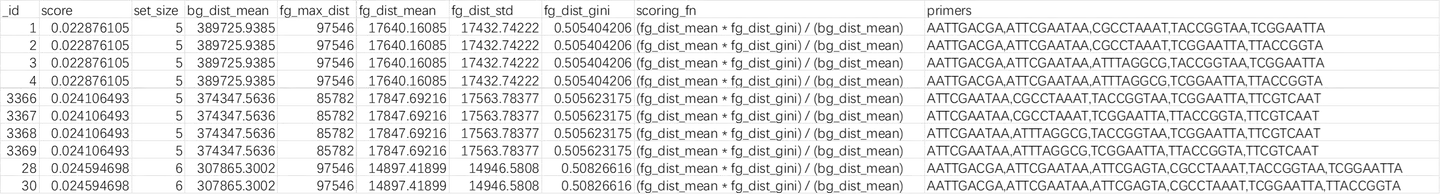
表格的每一行对应一个引物组。
参数说明:
score: 评分函数 (scoring_fn) 的结果。目前只有一个评分功能。
set_size: 集合中引物的数量。
bg_ratio: 背景数据集的长度除以背景数据集中引物结合位点的数量。这是一种计算背景数据集中引物结合位点之间平均距离的简单方法。
fg_max_dist: 目标数据集中任意两个引物结合位点之间的最大距离。
fg_dist_mean: 目标数据集中引物结合位点之间的平均距离。
fg_dist_std: 目标数据集中引物结合位点之间距离的标准偏差。
fg_dist_gini: 目标数据集中引物结合位点之间距离的基尼系数。基尼系数衡量数据集中的均匀性,其中 0 表示完全均匀(所有值都相同),1 表示尽可能不均匀。最大化引物覆盖的均匀性应该最大化扩增的均匀性。
scoring_fn: 根据我们当前的想法,根据当前评分函数 (fg_dist_mean * fg_dist_gini)) / (bg_ratio) 得出最低分数的引物是最佳的。这些引物组最小化目标基因组中引物结合位点之间的距离(fg_dist_mean),最小化基尼系数(fg_dist_gini:接近 0 的基尼系数值在整个目标基因组中分布最均匀),并最大化引物结合位点之间的距离背景数据集(bg_ratio)
primers: 引物组中引物的序列。
另外还可以总结目前的结果
在工作流程的任何时候,您都可以了解已收集的引物、已通过过滤器的引物以及已识别的集合。
$ swga summary输出将如下所示:
PRIMER SUMMARY
---------------
There are 379124 primers in the database.
0 are marked as active (i.e., they passed filter steps and
will be used to find sets of compatible primers.) Run `swga filter`
to identify primers to use.
The average number of foreground genome binding sites is 51.
(avg binding / genome_length = 0.000011)
The average number of background genome binding sites is 2800.
(avg binding / genome_length = 0.000001)
No melting temps have been calculated yet.
SETS SUMMARY
---------------
There are 0 sets in the database.
Run `swga find_sets` after identifying valid primers to begin
collecting sets.
---------------
Report generated from /Users/LAPT0011/swga_workspace/SWGA_FOLDER/primers.db

 浙公网安备 33010602011771号
浙公网安备 33010602011771号