03 CD-HIT基因聚类、去冗余
1、简介
CD-HIT是一个用于宏基因组分析的工具,它可以用于聚类和减少高通量测序(HTS)数据集的复杂性。在各种 HTS 平台上生成大量的序列数据,并对生物体中的微生物进行分类和注释时,该工具显得尤为重要。
在宏基因组分析中使用 CD-HIT,可以将高通量序列数据按照相似性信息进行分类,通过去除冗余序列来减少数据存储和计算复杂度。CD-HIT利用快速比对技术将非常相似的序列汇聚在一起,并生成一个代表性的序列集合。这样做可以在保留重要信息的同时,节省时间和计算资源。
在将 CD-HIT 应用到宏基因组分析中时,用户首先需要准备一个包含所有HTS Reads序列的FASTA文件,然后打开命令行终端窗口输入适当的参数,来运行CD-HIT。该工具还提供了许多其他选项,可以根据特定的需求进行定制,以便最大化其效果。
CD-HIT基因聚类、去冗余是一种用于蛋白质或核苷酸序列的聚类和比较软件,它可以利用贪婪算法和word过滤方法来快速、准确地找出相似性超过设定阈值的序列,并输出代表性序列和聚类信息。它在宏基因组分析中可以用于减少数据集复杂性、注释和发现微生物群落中的功能基因和蛋白质。
2、原理
CD-HIT是一种常用的聚类软件,广泛应用于宏基因组分析中。其原理如下:
-
序列预处理: 将输入序列进行长度排序,去除低质量的序列(默认阈值为50%),并删除冗余序列。
-
简化序列描述:由于同一个物种的不同基因可能在很大程度上重叠,所以使用一个较短的序列代表整个基因簇。
-
序列比较:对于每个输入序列,与前一个已经被选为代表的序列做BLAST比对,然后将相似性高于设定阈值(默认为90%)的序列加入该基因簇中,不断循环直到所有序列都寻找到了归属基因簇。
-
压缩基因簇:对于每个基因簇,选择具有最长序列的代表作为该基因簇的标记性序列,并扩展其它序列。
-
输出结果: 在输出文件中,包括每个基因簇的标记性序列、基因簇成员序列数量以及各基因簇之间的相似度等信息。
综上所述,CD-HIT算法通过BLAST比对和序列压缩的方法实现宏基因组数据的聚类,极大减少了输入序列的数量,并在较短时间内快速处理大量基因组学数据,从而实现高通量的宏基因组分析。
3、何为聚类、何为冗余
聚类是将具有相似特征的数据对象归类到同一组中的过程。在生物信息学中,聚类通常用于对DNA或RNA序列进行聚合和分类,以尽可能地减少基因集合的复杂性。
冗余是指存在重复或相似的信息。在生物信息学中,冗余通常是指两个或多个序列(例如蛋白质序列或核酸序列)之间存在高度相似或完全相同的区域。
举例说明,假设你有一个包含100个DNA序列的样本库,每个序列长度为1500 bp。使用CD-HIT对这些序列进行聚类,并减少复杂性后得到了50个非冗余的序列。这意味着在过去,你最初的100个序列中存在50个或更多的冗余区域。通过聚类和减少冗余部分,您现在只需要研究50个唯一序列,而不是100个。这大大简化了数据分析过程,提高了结果的可信度。
4、特点
CD-HIT是一种用于聚类分析的工具,它可以在大规模数据集中找到高度相似性的序列,并将它们组织成一个或多个簇。以下是CD-HIT的一些特点:
-
速度快:CD-HIT采用一种名为"word counting"的快速算法来比较序列相似性,因此能够快速处理大规模序列。
-
可扩展性好:CD-HIT支持多线程并行处理,可以运行在分布式计算机集群上,因此可以轻松地扩展到云计算平台上进行更大规模的分析。
-
精度高:CD-HIT使用了高度优化的蛋白质序列比对算法,并通过设置聚类阈值和去冗余策略来控制簇大小和精度。簇越小,则聚类越准确,但同时计算量也会增加。
-
支持多种文件格式:CD-HIT支持多种序列文件格式,例如FASTA、GenBank、EMBL等。这使得用户可以方便地导入自己的序列数据并进行聚类分析。
-
提供丰富的输出信息:CD-HIT可以生成包括聚类数量、序列数量、簇大小等在内的多种统计信息,以及每个聚类簇的代表序列,这使得进一步的序列分析变得更加容易。
5、输入文件与输出文件
CD-HIT 是一个有效的聚类算法,用于减小序列蛋白质数据集的大小。它可以基于多个序列对它们进行比较、分组并生成一份汇总报告。对于宏基因组分析,CD-HIT 是非常有用的工具。
输入文件:
- CD-HIT 的输入文件是 fasta 文件格式的一系列氨基酸序列文件或核苷酸序列文件(.fa 或 .fasta)。
- 输入文件应该由用户提供合理的阈值,以设置序列相似性和连接密度的界限。
- 如果您正在使用的是 CD-HIT-EST(特别用于 EST 数据的版本),则输入文件格式应为 .clstr 格式。
输出文件:
- CD-HIT 的输出文件命名规则是:输入文件名称_设定阈值_c序列名称.fasta/clstr
- c 序列名称是 CD-HIT 所选的每个群集中代表性序列的名称。
- 目标输出文件分为两个类型:结果报告文件 (.clstr) 和精简谷 (.fasta) 文件。
其中,结果报告文件 (clustering file, .clstr) 包含了这些信息:
- 每个簇中聚类的序列列表,以及代表性序列的名称、长度、所在簇的编号和在该簇中的编号。
- 每个簇的序列数目和代表性序列的长度。
- 基于 CD-HIT 的阈值,所产生的聚类数和不同聚类中代表性序列之间平均的精细度。
6、基本命令
cd-hit-est -i dna_all.fa -o out.fa -c 0.95 -G 0 -aS 0.9 -g 0 -M 0 -T 20
-i:输入文件名,dna_all.fa为fasta格式的DNA序列文件。-o:输出文件名,out.fa为输出的去冗余后的fasta格式的DNA序列文件。-c:相似度阈值(切比雪夫距离),本例中设为0.95,即两个序列相似度达到95%时被认为是同一个基因家族的成员。-G,-g:法定N/长序列限制。本例中都为0,即没有长度限制。-aS:全局序列比对参数。本例中设为0.9,即聚类时使用全局序列比对,且序列相似度需要达到90%以上才能归到同一簇中。-M:内存限制。本例中为0,即自动设置内存大小。-T:并行线程数。本例中为20,即同时使用20个CPU线程处理任务,提高处理速度。
###############################################################
conda install cd-hit -c bioconda
##cd-hit-est -i all2.fa -o out.fa -c 0.95 -G 0 -aS 0.9 -g 0 -M 0 -T 0
cd-hit-est -i dna_all.fa -o out.fa -c 0.95 -G 0 -aS 0.9 -g 0 -M 0 -T 20
perl out2_pro.pl ###筛选出对应的蛋白序列
################################################################
7、结果分析
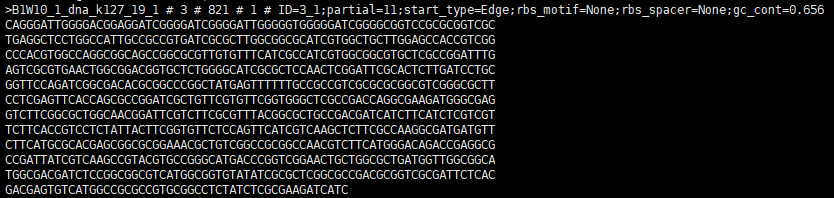
这是一个CD-HIT的聚类结果中的一行,其中各列代表的信息如下:
- 列1:B1W10_1_dna_k127_19_1,表示序列的名称或ID。
- 列2:3,表示该序列所在的簇或聚类中心的标识符。
- 列3:821,表示该序列的长度。
- 列4:1,表示在该簇中的序列数量,本例中为1个。
- 列5:ID=3_1;partial=11;start_type=Edge;rbs_motif=None;rbs_spacer=None;gc_cont=0.656,表示序列的附加信息,以分号分隔。
- ID=3_1:表示该序列在原始FASTA文件中的ID。
- partial=11:表示该序列为其原始序列的部分序列(partial),且起始位置为第11个碱基。
- start_type=Edge:表示该序列相对于整个基因组的位置为边缘(Edge)。
- rbs_motif=None;rbs_spacer=None:表示该序列中的可能的启动子或RBS序列(rbs_motif)和其Spacer序列(rbs_spacer)不存在。
- gc_cont=0.656:表示该序列的GC含量为0.656。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号