
关于RDkit与PDB中小分子的迷思
目的:对于既含有蛋白质,也含有非天然残基、小分子、离子的PDB,提取分子SMILES和原子级别的特征
1. 首先,我考虑了直接用Rdkit来处理完整PDB
直接读取pdb生成rdkit.mol对象: mol = Chem.MolFromPDBFile(input_pdb)
Chem.SplitMolByPDBChainId可以将每个chain划分为一个mol,返回{chain_id: 这条chain对应的mol对象}字典:
chain_mols = Chem.SplitMolByPDBChainId(mol)
for chain_id, chain_mol in chain_mols:
xxxxxxxxx
Chem.SplitMolByResname(mol)按照resname来划分小分子,返回:{resname: 这个res对应的mol对象}字典
因为是字典,可想而知,对于多个相同的resname,比如蛋白质中重复出现的氨基酸,后面残基的mol对象就会覆盖前面的
如果还额外输入了rename的列表,如Chem.SplitMolByResname(mol, [target_resname]),那么返回的字典里面就只会包含这些resname
理想很丰满,现实很骨感。RDkit的许多操作都需要mol通过分子校验sanitize,否则无法进行或无法正确进行诸如RemoveHs, MolToSmiles和各种Split划分submol的从操作,然而PDB文件经常缺侧链少原子,导致十有八九RDkit都处理不了报错
于是,我又依次尝试了如下方法:
- 使用Pymol从原始PDB中专门提出非天然残基或小分子resname对应的部分,形成uaa.pdb,然后RDkit读取处理:
情况比读整个PDB好一些,因为只要uaa部分过了sanitize就可以被RDkit读入,但是出现了新问题:sanitize过程中会推断H的连接,导致一些正离子如Ca2+变成了[CaH2]
- 提取uaa.pdb → openbabel转成小分子的mol2格式
obabel -ipdb uaa.pdb -omol2 -O uaa.mol2
.mol2会自动根据输入的pdb推断原子电荷,由于uaa.pdb里面只有重原子,所以最后产生的mol2被rdkit读入后带有一堆不合理的电荷或自由基
![]()
- 提取uaa.pdb → openbabel转换格式的时候使用-h参数自动推断补H
obabel -ipdb uaa.pdb -omol2 -O uaa.addh.mol2 -h
会出现和直接使用rdkit读取uaa.pdb一样的问题,即补H也会让Ca2+变成[CaH2]
- 提取uaa.pdb → Pymol命令
cmd.h_add加H →cmd.save保存为uaa.addh.pdb → openbabel转成mol2obabel -ipdb uaa.addh.pdb -omol2 -O uaa.addh.mol2
只要提前补好了H,并且不指定-h参数,obabel就会根据已有的H来推断重原子电荷,基本上是正确的。但是这时我发现了第二个问题:
4.1 从Pymol输出的PDB文件会丢失原始的,由CONECT字段定义的不饱和键连接信息,因此从Pymol中输出的uaa.pdb里面的不饱和键已经丢失了,在此基础上补的H完全是错误的!后面就一步错步步错了!
解决:所有从PDB文件里面提取子结构的操作均换用biopandas处理,输出时保证ppdb.df['OTEHRS']里面'record_name'=='CONECT'的部分一并输出
4.2 cmd.h_add这一步实在无法规避Pymol,虽然补H的时候是正确的,但是输出文件又会丢失不饱和键信息,导致obabel的结构和电荷推断错误
解决:对于Pymol输出的uaa.addh.pdb,手动从biopandas生成的uaa.pdb中把所有的CONECT行全部复制过来加到末尾,这一步需要注意在使用Pymol时设置cmd.set('pdb_restain_ids', 1),否则Pymol会将所有原子重新从1开始编号,这样从uaa.pdb中复制过来的CONECT中记录键两端的原子序数就和uaa.addh.pdb里的原子序数对不上了
(我也尝试过PDBFixer能否补H,结果发现PDBFixer不识别非天然残基和小分子,并且会在输出文件中将他们直接删除!)
以上,分子本身的问题基本解决了,但是这时候发现了RDkit的笨比sanitize又有新问题:
0. 首先注意:Chem.MolFromMol2File(input_mol2)生成的mol无法再Chem.SplitMolByResname(mol),尽管mol2文件里面含有原子所属分子名称和序号的信息
关于mol2格式的具体介绍,参考:谈谈记录化学体系结构的mol2文件 - 思想家公社的门口:量子化学·分子模拟·二次元
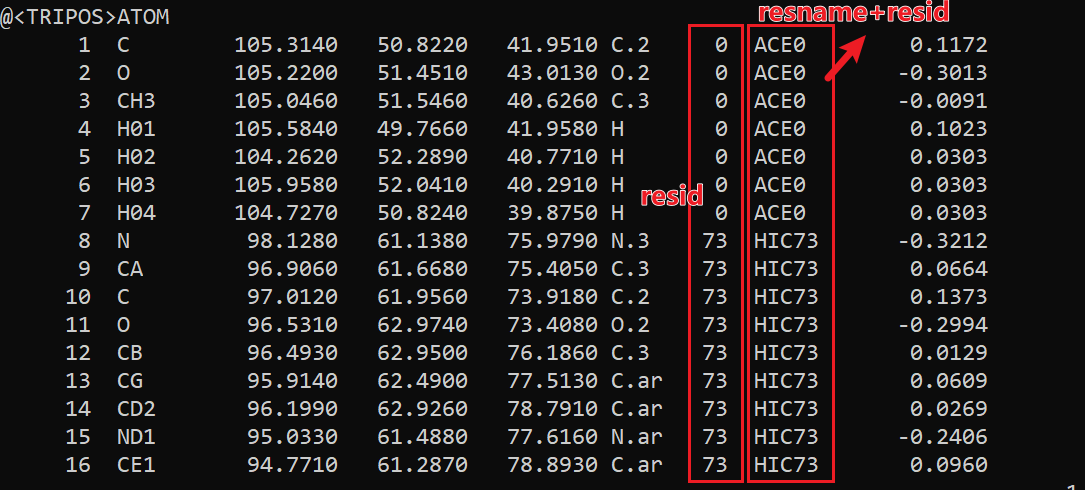
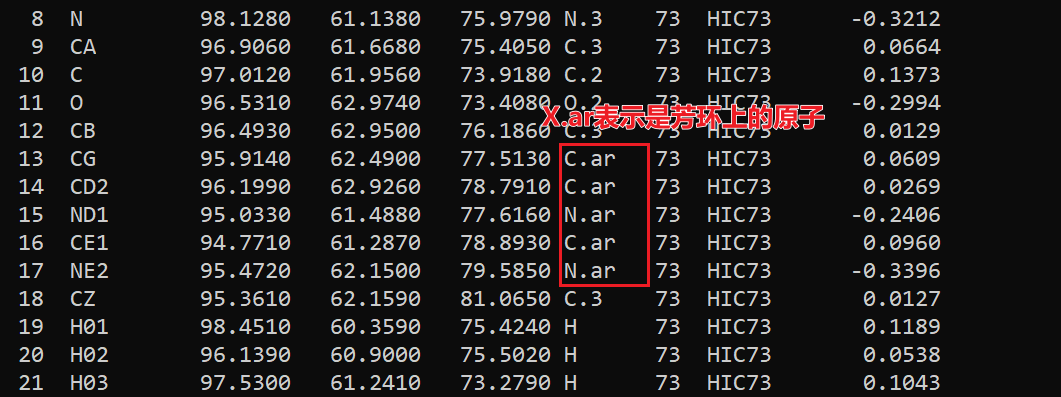
这时候只能使用submols = Chem.GetMolFrags(mol, asMols=True)来拆分每一个小分子(相当于把SMILES按照非键分隔符.断开),在后处理中人工匹配resname和submol
- RDkit的sanitize强行要求将芳环表示为凯库勒式(单双键交替的形式),而.mol2的芳环原子是X.Ar表示,芳环的键均使用ar表示,由于笨比RDkit不会推断H的连接方式进而将大π键转化为单双键交替形式,所以几乎含N杂环(比如我遇到的咪唑)sanitize必出错,官方也说了这个问题确实没法解决:mark this one as "won't fix"
![]()
![]()

Can't kekulize mol · Issue #7343 · rdkit/rdkit
FrequentlyAskedQuestions - cant-kekulize-mol · rdkit/rdkit Wiki
搜索发现某个issue里面建议用.mol而不是.mol2(但是具体是哪个页面因为当时搜了太多找不到了),因为.mol本身就是用单双键交替来表示芳香键的,天然与RDkit的sanitize兼容,经验证这种方式在atom.GetIsAromatic()输出也是正确的。
obabel -ipdb uaa.addh.pdb -omol -O uaa.addh.mol,然后Chem.MolFromMolFile(input_mol)
但是!.mol也有问题:虽然基本都能通过sanitize,但是读取出来的mol却不含正确的形式电荷信息(或者是openbabel在生成.mol的时候不会自动推断电荷信息),比如[Ca2+]会变成[Ca],本来该带电的N也不带电了,这样Atom.GetFormalCharge()就得不到我想要的结果了
-
进一步发现,Pymol在面对酸根离子如SO42-或者PO43-的时候也会把它们加氢成不带电的酸根,此外,一些本来应该是二价的离子如Zn2+和Cd2+最后不知道为什么得到的确是[Zn-]和[Cd-]
解决:加H这一步不用Pymol,换用Chimera,不会给酸根离子加H。虽然Chimera的write命令可以选择直接输出mol2文件write format mol2 #0 output_filename,但是发现chimera直接输出的mol2连接关系有问题,似乎是它不会像openbabel那样智能地按照实际原子连接关系来推断电荷,而是默认了一些原子之间的键的类型
![]()
-
chimera的问题:chimera加H后write PDB会画蛇添足地加CONNECT,不同的小分子之间连起来
![]()
此外,chimera不会给N-C-C=O(即alpha氨基酸骨架的C=O)补H,这会导致openbabel转化时的错误?

最后的解决方法是:
biopandas从原始pdb里面提取uaa.pdb → Pymol给uaa.pdb加H生成uaa.addh.pdb → 把uaa.pdb里面的CONECT字段补充到uaa.addh.pdb里面 → openbabel将uaa.addh.pdb转化成mol2 + mol = Chem.MolFromMol2File → 如果sanitize失败即mol = None,再用openbabel将uaa.addh.pdb转成mol + mol = Chem.MolFromMolFile
当然这个pipeline还有一些细节问题:如果是个有N端和C端的非天然残基,原始PDB里也不会包含其肽键C=O是个双键的连接信息,这样Pymol读入的uaa.pdb的时候会自动把这里当成单键的C-O,然后补H的时候会把这里补成-COH,最后导致得到的SMILES不正确;如果.mol2用不了不得不用.mol,得不到正确的电荷信息,我实在是不想搞!就这样吧!
总的来说要注意:
1.提取步:pymol会导致不饱和键的连接信息丢失
所以,能用biopandas用biopandas,实在规避不了pymol,需要从原PDB中复制CONECT行过来补在pymol输出的pdb文件末尾
并且使用Pymol时首先cmd.set('pdb_retain_ids', '1'),否则Pymol输出的pdb会将原子从1开始重新编号,这样即使从原PDB中复制CONECT行过来,键的atom_idx也对不上了
2. openbabel步:
使用pymol加H并补充了正确CONECT连接信息的.pdb作为输入,不要使用obabel -h自动推断补H,会让Ca2+变成[CaH2]
3. RDkit步:
.mol比.mol2更容易通过sanitize,但是.mol不含有电荷信息,导致Ca2+变成[Ca]
.mol2信息最完整,但是一旦设计含N杂环几乎必然通不过sanitize,因为RDkit的sanitize要将分子转化为凯库勒式,但是笨比RDkit又不会推断H的连接

 浙公网安备 33010602011771号
浙公网安备 33010602011771号