
8种特殊建库测序
8种特殊建库测序
1. RNA-seq
2. 外显子测序
3. small RNA-seq
4. 单细胞DNA测序
5. 单细胞mRNA测序
6. 甲基化测序
7.Moleculo长测序
8. Ribozero和方向性RNA文库
1. RNA-seq
今天呐,主要是给大家介绍一下RNA-seq(RNAsequencing)。也就是RNA的高通量测序技术。
因为我们这个节目主要是针对医学方面的技术进展,所以,我们今天对于RNA的介绍,也会侧重于人的RNA测序方面的技术介绍,以及相关的生物信息学方面的工作。
RNA高通量测序(RNA-sequencing,缩写为RNA-seq)是目前高通量测序技术中被用得最广的
一种技术,RNA-seq可以帮助我们了解:各种比较条件下,所有基因的表达情况的差异。
它可以检测的差异有:正常组织和肿瘤组织的之间的差异;它呐,也可以检测药物治疗前后,基因表达的差异;它呐还可以检测发育过程中,不同的发育阶段,不同的组织之间的基因表达差异。诸如此类呐,很多。我们不一一列举。
那么在所有检测的差异类型中,最常见的,就是检测所有mRNA的表达量的差异,这是最常用的一种检测。
同时呐,我们还可以检测 RNA 的结构上的差异。例如:mRNA的剪接方式的差异,也就是我们一般说的“可变剪接”,还可以检测“融合基因”,同时还可以检测基因单点突变导致的SNP(Single Nucleotide Polymorphisom)。
接下来,我们分成“RNA-seq测序方法”和“RNA-seq测序数据分析”两个部分,分别介绍RNA-seq。
RNA测序方法
在测mRNA的过程当中,首先要解决的问题,是如何去除核糖体RNA也就是去除“rRNA”(Ribosomal RNA)。
那在通常抽提到的总RNA中,绝大部分都是核糖体RNA(rRNA)。以人类的细胞或组织为例,一般抽提到的总RNA当中,95%都是核糖体RNA。剩下的2%到3%是mRNA。还有呐,2%到3%是Long non-coding RNA、或者tRNA、microRNA,这些RNA,也就是说mRNA只占了所有RNA中的一小部分。
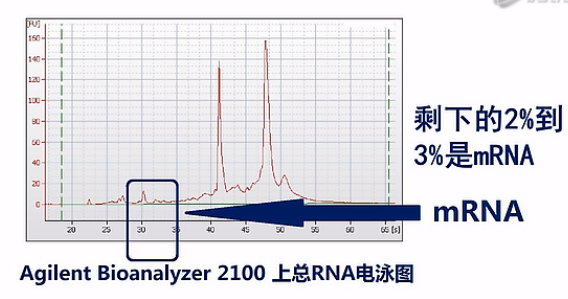
如果我们把所有的RNA都拿来测序的话呐,测到的绝大部分的序列数据呐,都是核糖体RNA。而且这当中(rRNA)比例会高达95%左右,但是呐,核糖体RNA在整个人类当中都是非常保守的,而且在人的各个组织、器官当中也是极度稳定的。也就是说,测rRNA,它得到的数据,并不能为我们实验者提供什么有用的信息,而mRNA才是RNA当中信息含量最丰富的那个部分。
我们一般的RNA-seq要测的,也是mRNA的各种变化,所以呐,在实验过程当中,我们一般要把核糖体RNA先去掉。然后再进行建库测序。
去除核糖体RNA,并进行建库的方法,有许多种。
今天呐,我们主要介绍一下应用最广泛的illumina公司的TruseqRNA建库方法。其它的方法呐,以后我们再找机会再给大家做介绍。
那么这张呐,就是mRNA测序的建库过程图。首先呐,是利用高等生物的mRNA都有Poly(A)尾巴这个特点,用带有Poly(T)探针的磁珠与总RNA进行杂交。然后呐,Poly(T)探针呐,就和带Poly(A)尾巴的mRNA结合在一起,接下来呐就回收磁珠,然后呐把这些带Poly(A)的mRNA从磁珠上洗脱下来。
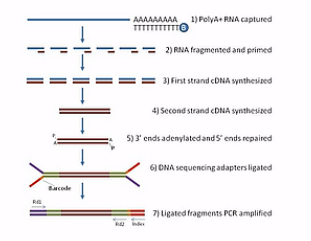

然后呐,再把这些洗脱下来的mRNA用镁离子溶液进行处理。镁离子溶液会把mRNA打断。

被打断的这些mRNA片段,再用随机引物进行逆转录。

逆转录成(第一链)cDNA后,再合成出第二链(cDNA)。这样就成为双链的cDNA。接下来呐,我们再在双链的cDNA的两端接上“Y”型的接头。这样呐,就成了标准的测序文库,然后呐,这个标准的测序文库就可以拿到HiSeq测序仪上进行测序了。



样本质量要求
在这里呐要说明一下,这个建库方法对RNA的完整度有较高的要求。也就是说,只有在mRNA大部分是完整的状态下,才能得到比较好的效果。
这是因为带Poly(T)的磁珠,它所吸附的是Poly(A)的那些序列。那么如果mRNA发生了降解,也就是mRNA断掉了,那么磁珠所吸附下来的片段,都是那些靠近3'端的那些断片,而那些5'端的断片呢,是吸附不下来的。会在富集过程中被洗脱掉。
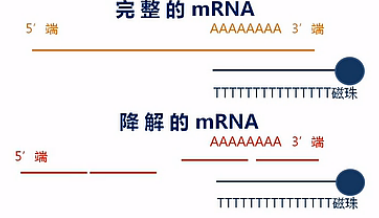
那么这样呐,接下来的数据分析当中,就会发生一定的数据偏差。
那么为了保证能够测到尽可能完整的mRNA序列呢,Illumina公司是这样建议的:它建议先对总RNA进行一次质量检测,一般是用Agilent公司出品的Bioanalyzer 2100毛细管电泳仪,对总RNA样本进行一次电泳质检。那Bioanalyzer呐会根据18S和28S这两个核糖体RNA的电泳峰是否高、是否尖,来判断RNA的质量。并且呐,会自动打分。
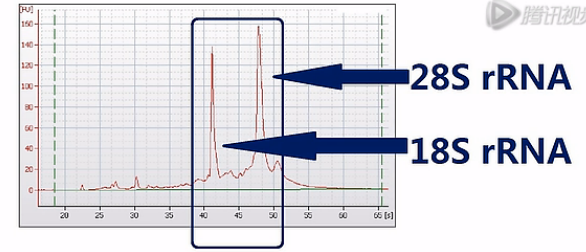
这两个峰越高、越尖,也就说明RNA的降解就越少,完整度呐就越高。那么打分呐,也会越高。反之呐,打分就会低。这个分值呐,叫“RIN”值。也就是RNA的完整度评分值。是“RNA Integrity Number”的英文首字母缩写。RIN值最高是10分,最低呐是0分。
Illumina公司推荐用RIN值在8.0以上的RNA进行建库和测序。测序完成之后呐,就可以进行数据分析了。
数据分析
分析的第一步呐,一般是先把测到的RNA片段,先mapping(比对)到基因组上,那么在比对完了之后呐,可以先看一下,有多少的RNA片段,是在靠近基因的5'端的位置,又有多少片段在是靠近基因的3'端的位置。
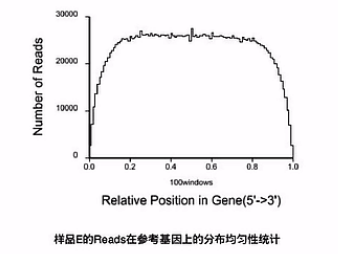

那么这张图上呐,就是把所有的基因,都按其外显子的长度呐,拉直,然后呐,归一化到“0 - 100”的这样一个长度。然后呐来看,比对上的片段,有多少是落在这0到100的这一个轴的哪个位置上。
这样一个比对的结果,就可以让我们看见前面Poly(T)磁珠在抓mRNA的时侯。
捕获下来的这些mRNA是不是完整的,如果捕获下来的这些mRNA大部分是完整的话呐,那么这个图形靠近5'端的曲线就会显得比较饱满。它的高度会和3'端的高度差不多。
反之,如果这根曲线的3'端是很高的,而5'端是比较低的,我们就可以初步判断,这个RNA有一定程度的降解。
因此,我们可以推断在捕获过程当中,有相当一部分(mRNA),它的5'片段因为与3'片段的Poly(A)片段的尾巴断开了,所以,没有被捕获下来。所以,这个RNA呐,是有一定程度降解的。
在知道了测序的质量之后呐,接下大家来要关注的就是不同样本之间、各个基因的mRNA的表达量的差异。
RPKM 指标
那么在做这些比较的过程当中,目前最常用的,对基因表达量进行相对定量的一个指标,就是RPKM值。那么RPKM呐,是Reads Per Kilobase of exon model perMillion mapped reads的英文的首字母缩写。
RPKM翻译成中文呐,就是每一百万条可以比对到基因组上的Read当中,有几条是可以比对到某个特定基因的,
然后呐这数值再除以该基因的外显子的长度,得到的这样一个最终的比值。

这个公式呐,就是这样的。
它的分子呐,就是经对到某个基因的外显子的read数。它的分母的第一项呐,就是这次所有比对到基因组上的read数(M reads,MillionReads)。分母的第二项,就是这个特定基因的外显子的长度。
我们接下来分步地对这个公式进行一下解释,首先呐,就是比对到某个基因的外显子上
的Read数,去除以这次所测到的、全部可以比对到基因组上的Read数。这个比较容易理解就是:这个基因所表达出来的mRNA,它所被测到的片段,来和所有被测到的、可以Mapping(比对)到基因组上的片段来进行比较。这点是比较容易理解的。
那么这个比较费解的是,为什么还要除以第二项,就是“除以这个外显子的长度”。这是因为建库过程当中,这个RNA是用镁离子溶液来处理,
然后打断(并逆录)成若干个180-200BP左右的小片段,如果一个基因的长显子越长,那么它所产生的mRNA就越长,那么mRNA越长呐,被打出来的小片段就越多。
我们来假设,一个A基因,它的mRNA的长度呐,假设它是1Kb,那么它的1Kb的mRNA可能被打成“5”个,200Bp左右的小片段;那么还有一个B基因,如果这个B基因的mRNA是2Kb长,那么,它同样被打成200Bp左右的小片段呐,它就会产生“10”个小片段。
我们来看,A基因是5个小片段,而B基因是整整10个小片段,所以,B基因在测序过程当中,它被测到的概率就会比A基因整整大出去一倍。
这就是我们为什么要把刚才第一项比出来的比值呐,然后再除以这个外显子的长度。
通过上面的解释呐,我们就可以理解:除以这个外显子的长度,它的目的:是修正这个mRNA长度所引起的mRNA的Read数的偏差。
通过这种修正呐,能够还原出一个比较真实的、原始的表达拷贝数状态。
这个呐,就是“RPKM”定义的原理。
火山图
那么作为一种针对全转录组的分析,我们希望是一次看到一个整体的样本(表达)差异的情况。而不仅仅是看少数几个基因的表达差异。
科学家做了一种叫“火山图”的一个图形,来比较形象地来说明2个样本之间的表达差异。
那么我们来看这张图,这张图呐,这个样子就象火山喷发的样子,那么这个图呐,是2个样本的RNA的表达量的对比。
这个图的横轴呐,是表示某个基因的表达是上升了,还是下降了。
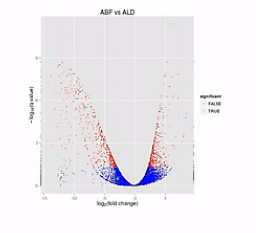
纵轴是表示这种差异的置信程度,这其中的每个点呐,就是两个样本当中同一个基因的mRNA表达量的变化。
如果这个基因的表达是上调了,那么这个点呐,就往右移动。反之,如果这个基因的表达量是下调了,那么这个点呐,就往原点的左移动。
那么这个纵轴呐,就是这种变化差异的置信程度。如果这个置信程度越高呐,那么这个点的纵轴位置也越高。
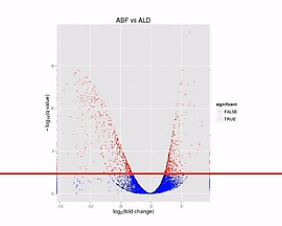
那么我们在纵轴上划了这样一条水平线,超过这个水平线以上的(点)呐,(其差异水平的)置信程度是很高的。我们就把它标示成红颜色。如果低于(这条水平线的)置信程度呐,它的置信程度也相对低一些,我们把它标成蓝颜色。
这里要解释一下,为什么差异程度是相同的情况下,它们的差异置信程度是不一样的。比如说同样是差了2的5次方,也就是32倍,它的差异置信程度会不一样,有些是蓝点,有些是红点。

A基因在甲样本中,被测到了3200条,而在乙样本中被测到了100条;B基因在甲样本中,被测到了320条,而在乙样本中被测到了10条。它们同样是差了31倍,但是因为A基因的样本统计数,远大于B基因的样本统计数,也就是说,它们的Reads数有那么大的差距。
所以,A基因的这个差异的置信程度,会比B基因的这个差异置信程度要高许多。
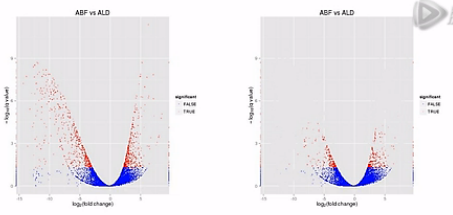
那么,我们再来对比这两张图。那么就可以比较直观地发觉,左侧的这个图当中,有更多的基因表现出明显的差异,这样呐,火山图就为我们提供了一个形象的、直观的、整体表达差异信息。
聚类分析图
聚类分析呐,是RNA分析中非常常用的一个手段。它呐是通过多个样本的全基因表达谱对比,
来找到它们之间的相似性,和相近关系。
这是一张聚类分析的图,横轴呐是样本,纵轴呐是基因。通过聚类分析呐,可以发现:在这个群体中,样本被分成了3个群体。
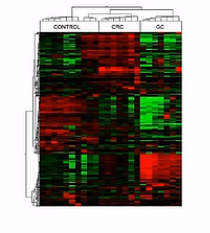
每个群体的内部呐,都有着相似的表达特征。同时,我们还可以看到,基因的表达,也是成簇的,这儿呐大体上分成3个基因群。那么这3个基因群呐,各自有着相似的表达量。
聚类分析呐,有很多的应用,比如说:我们可以分析疾病的亚型。
那么还可以通过对多个基因在特定疾病当中的表达倾向性呐,来找出可能的、新的、诊断用的Biomark。
聚类分析,有很多新的应用,有待我们一一去开发。
GO分析
GO分析是RNA-seq分析中非常常用的一种分析。GO是Gene Ontology的缩写,GeneOntology呐是一个国际化的、基因功能分类体系。这个体系用一整套动态更新的标准词汇、和严格定义的概念,来全面地概括任何生物中基因和基因产物的属性。
GO主要描述基因的三个属性:
第一,是这个基因,它参与的生物过程
第二,是这个基因的产物的功能
第三、是这个基因产物在细胞器内的空间定位
差异基因GO富集柱状图:可以直观的反映出在生物过程、细胞组分、和分子功能富集的差异基因的个数分布情况。
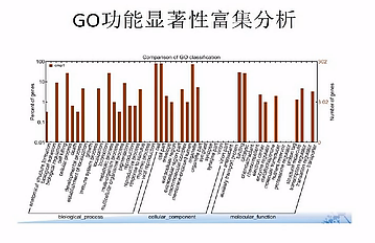

有向无环图,是差异基因GO富集分析的图形化展示方式,从上到下呐,它所定义的功能范围越来越小、越来越精准。
它的分支呐,表示包含关系。而这个圈圈的颜色越深呐,表示这个富集关系程度越高。
Pathway分析
通路分析:通路(Pathway)是指在系统水平上完成生物的某一功能的基本单元、或者局部子网络。
KEGG,也就是:Kyoto Encyclopaedia of Genes andGenomes。翻成中文:就是《京都基因和基因组百科全书》,是目前公认的、最权威的基因功能数据库。
这其中的Pathway(通路)是KEGG的核心内容。
目前针对Pathway的分析、注释,大多数是基于KEGGPathway来做的。
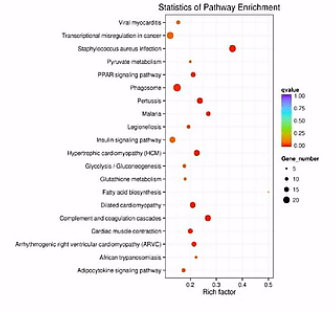
散点图是KEGG富集分析结果的图形化展示方式。
在此图中,KEGG富集程度通过Rich factor、Qvalue和富集到此通路上的基因个数来衡量。
点的面积越大呐,则富集的基因数越多。
富集的因子越大呐,则表示富集的程度越大。
qValue呐,是校正之后的pValue。那么它越接近于0呐,表示富集程度越显著。
结构变异分析
前面,我们讲的都是基于RNA表达量的差异分析。
接下来呐,我们要说一下,RNA-seq当中,可以测到的mRNA上的各种结构上的变异。
所谓结构上的变异呐,也就是RNA序列的变异。
主要呐,是3种:
第1种,是可变剪接
第2种呐,是融合基因
第3种呐,是点突变,也就是SNP
结构分析需要较深的测序深度
这里要说明一下,对于想要测mRNA结构变异的用户呢,建议测序深度要测比较深。
我们一般呐是建议测10G以上的数据量。那么为什么要测这么多的数据量呐?原因是二代测序,目前的测长还不是很长,每一个Read呐,只有大约100到125个Bp左右。如果测序深度不够呐,那么读到的这些read在整个的mRNA上的分布呐,是一种比较零碎的一种状态。
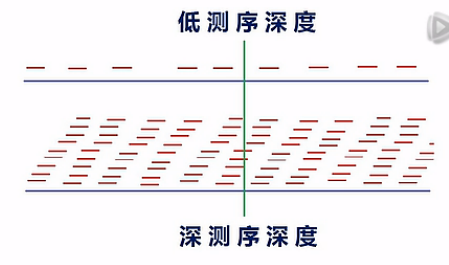
那么在这种比较零碎的、不完整的覆盖情况下,要去分析哪里有一个剪接点,哪里有一个断点,哪里有一个SNP,它不是很准确的。
当测序深度足够深的时侯,在每一个位点,都有10几次、或者几10次的覆盖的时侯呐,我们就可以比较有把握地来判断出,哪儿有了一个新的剪接点,哪儿出现了一个断点,哪儿,碱基发生了突变。
可变剪接
可变剪接,在真核生物中普通存在。一般一个人的组织样本当中呐,可以通过高通量测序,发现有5000个到20000个左右的可变剪接。
融合基因

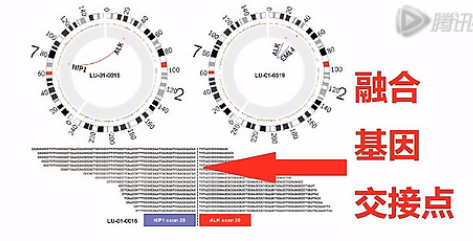
融合基因呐,是指原来在基因组上分开的2个基因,因为某种原因,染色体发生了重排。
重排的结果呐,是让A基因的头,接到了B基因的身体上,这样就产生了融合基因。
那么这张图呐,就是一个癌细胞中的融合基因的示意图。
接下来这张图呐,是高通量测序测到融合基因的这个图。我们可以看到这10几个Reads都横跨在这个融合基因的、交接点的两侧,由此呐,证明了这个癌细胞当中有这么一个融合基因。
点突变

RNA-seq呐,还可以找出点突变,这个呐,是一张泡泡图,来表示我们所找到的点突变。
发生突变频率最高的这个基因,就用最大的泡泡来表示。(突变)频率低一点的,就画一个小一点的泡泡(频率),再小一点,那么再小一点的泡泡。
这些泡泡呈逆时针排列,形成这样一个泡泡图。
2. 外显子测序
今天,我们会和大家谈一下人外显子组测序的方法原理。和它能够给我们带来哪些有用的生物信息。
那我们还是分两个部分来介绍,第一个部分呐,介绍外显子测序的技术方法。第二个部分,我们来介绍外显子测序可以得到哪些有用的生物信息。
实验原理
那么,我们先来说外显子测序的工作原理。
我们先来看这张图,外显子测序的核心技术呐,是这(些)个针对人外显子序列设计的捕获探针库,那么这些探针的序列呐,都和人外显子的DNA序列相互补。
在实验过程中呐,它可和人的外显子DNA序列进行杂交结合。同时呐,这些探针都标上了生物素。
有了这个捕获试剂盒呐,就可以进行建库、和捕获了。
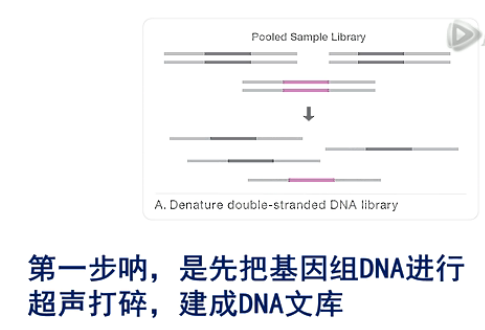
第一步呐,是先把基因组DNA进行超声打碎,建成DNA文库。
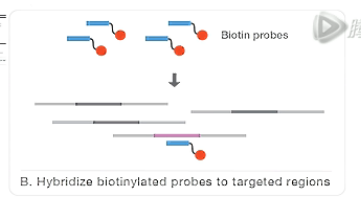
第二步呐,是把建好的文库和探针库进行杂交。
杂交过程中,通过核酸序列的互补结合的原理,探针会和目标DNA片段进行结合。
然后呐,再用结合了链霉亲和素的磁珠,与这个杂交混合液呐进行混合。
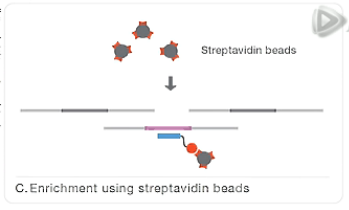
因为链霉亲合素是会和生物素牢固结合的。
这样,就把我们要捕获的外显子目标片段,通过探针,间接地结合到了磁珠上。
然后呐,通过磁铁把这些磁珠给吸附下来。
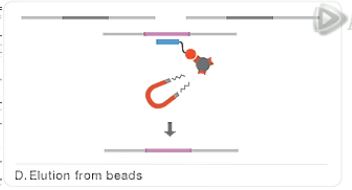
而把上清液呐给去掉。
这样呐,也就把没有结合的DNA片段给洗掉了。
再接下来,用洗脱液,把我们要的DNA文库从磁珠上给洗脱下来。
那么这些文库呐,再经过PCR扩增,就可以上HiSeq测序仪进行测序了。
测完序以后,就可以进行生物信息学的处理了。
数据分析
那么先是把这些测序的DNA片段比对到人的基因组上。
然后呐,把这些比对到基因组的序列进行突变分析。
覆盖深度
一般用Agilent SureSelect 50M的试剂盒进行外显子建库、捕获。再用HiSeq 2500 V4 PE125的方法进行测序,测10个G的数据量。大约可以得到95X的“有效测序深度”。我们说的有效测序深度(effective sequencing depth)是相对于总测序深度来说的。
总的测序深度呐,是把所有测得的数据(量)去除以目标区域的大小。
举例来说,用Agilent 50M的这个试剂盒,我们测到10个G的数据。
去除以目标区域的50M的大小,那么得到的是200X的测序深度(10G/50M = 200X)。
但是这个200X的测序深度,对于做生物信息学分析来说呐,并没有太大的实在意义。
因为当中还要扣掉许多无用的数据,才能得到有效的数据。
在外显子测序中,要扣掉4种因素引起的无效数据。
第一个影响因素呐,就是因为杂交捕获的过程它不是十分精确的,基因组中的有许多序列,
是和外显子有一定的同源性的。那么这些片段呐,在杂交过程当中,也会被杂交捕获下来。而这些片段呐,不是基因的外显子。

所以,我们在分析过程当中,首先要把这些序列给去除掉。
第二个影响因素呐,是捕获下来的一个片段,很可能它(只有)一部分的序列是落在目标区域还有一部分序列呐,是突出在目标范围之外的。那么,这部分突出来的序列呐,它不是目标区域,所以,它也不计入外显子测序的“有效测序深度”。
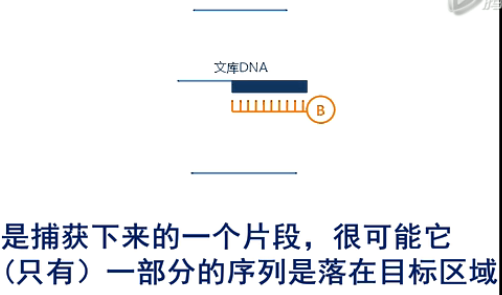

这个落在目标区的数据,占全部被测到的数据的比例,我们用一个专业术语来称乎它,叫作
“捕获效率”(capture efficiency)。那么AgilentSureSelect这个试剂盒呐,它的捕获效率,大约是65~70%。
第3个影响有效数据比例的因素呐,是Duplication。那么所谓duplication呐,就是建库过程当中它的最后一步,是通过PCR扩增把原始的模板,扩增出几百倍来。那么由同一个模板分子扩增出来的子文库分子呐,是长得一模一样的那么这些多出来的这些分子,如果被重复地测到它并不能为我们提供太多有用的生物信息。所以,我们在生物信息分析的过程当中,
我们要去掉这些重复的片段。
那么要去掉这些重复的片段我们判断的依据是:2个DNA分子它们的5'起始位置、和3'的结束位置,完全一模一样,那么我们就认定这2个分子,是从同一个母分子,PCR出来的(2个)子分子。
然后呐,我们会比较这2个序列的数据质量,留下那个数据质量比较高的,去掉那个
数据质量比较低的,这也就是我们通常所说的“去Duplication"的过程。
用Agilent SureSelect试剂盒进行建库、捕获,实测10个G的数据,我们发现duplication大约在5%左右。
那么,我们说明一下,duplication的比例不是恒定不变的。而是会随着测序深度的增加、而增加,因为上机(测序)的文库是经过PCR扩增的文库。
随着测序量增大,那么测到源自同一个模板的PCR子分子的概率呐,就会提高。
第4个影响因素呐,是目前主流的测序方法是HiSeq V4 PE125这种方法。也就是:双端各测125个碱基,那么Agilent的建库方法中当呐,插入片段是150~200BP,这样一个大致范围的这些片段,那么它的平均片段长度呐,是180BP。那么我们用双端125的方法来测序就会导致左边的这个reads(序列)和右边的这个reads(序列),读到当中,会有一段,大概会有70BP的交叠。
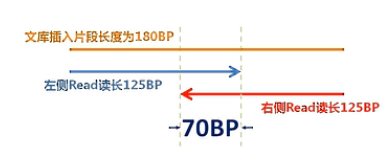
那这个70BP的交叠的序列,是冗余的序列。也就是说,我们读了250个BP的序列,但是
其中大约有效的呢,是180个(BP)。有70个BP呐,是冗余的。
综合上述4项因素,我们可以看到,用AgilentSureSelect 50M的这个捕获试剂盒进行建库、捕获,并且用HiSeq V4 PE125的测序方法来测序,测10个G的数据量,那么可以得到,大约95X的有效测序深度,(10 * 0.7 * 0.95 * 180 / 250 / 50 = 95)。
覆盖均匀性
除了测到的有效数据量之外,还有一个因素会影响到后面的分析,这就是Reads的分布均匀情况,也就是说目标区域的每个碱基被覆盖的深度的均匀性。那么这个结果呐,是越均匀越好。

科学家经过实测,发现Agilent的SureSelect、和Roche的Nimblegen,这两个捕获试剂盒,所得到的覆盖均匀性是比较好的。
在肿瘤测序中的优势
外显子测序,可以测Germline突变(胚胎形成时就带有的突变),也可以测体细胞突变(Somatic Mutation),但是呐,随着Illumina推出HiSeq X10测序仪,把人全基因组测序的直接成本降到1000美元以下,那么Germline水平的突变呐,已经很少用外显子来测了。
目前,外显子测序的主要优势就体现在肿瘤基因测序方面,之所以外显子测序在肿瘤基因测序方面有优势呐,这是因为外显子测序,它的测序深度,可以比较容易地做到“深度”测序。那么呐,它可以比较轻易地达到100X、200X,甚至更深的测序深度。这个呐,就有利于测到 low allele frequency (低等位基因频名优新)的体细胞突变。
因为肿瘤中的突变呐,往往都是 low allele frequency 的体细胞突变。所以,外显子组测序就在测肿瘤基因组突变方面,显出比较明显的优势来。
那么如果是要测肿瘤中的体细胞突变呐,一般是拿手术切下来的肿瘤组织DNA、和病人外周血中的白细胞基因组DNA,进行外显子测序。
一般肿瘤的测100~200X的深度,白细胞的(DNA)测100X的深度。从白细胞DNA得到这个病人的Germline基因组序列,拿肿瘤的DNA序列与之做对比,找出其中的体细胞突变。
SNP信息
外显子组测序,主要能够得到的信息是点突变,也就是SNP信息,和插入缺失突变,也就是Indel信息。这张图呐,就是找到的体细胞突变的泡泡图。
在这个泡泡图中,突变频率越高的基因,就画一个大泡泡,放在图的中间。突变频率低一点的基因,就画一个相对小一点的泡泡,延着逆时针排列。再低频的突变,再画一个再小的泡泡,再在外面再排列下去。依此类推,就得到这样一个泡泡图。
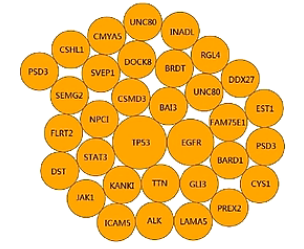
泡泡图有利于我们非常直观地看到样本中体细胞突变的情况。
GO 和 Pathway 分析
找到突变之后,就可以进一步地做GO和Pathway分析。
关于GO和Pathway的定义,我们已经在前一期讲RNA-seq的节目当中,给大家介绍过了,想要了解的朋友可以在优酷中找一下:【陈巍学基因】视频7:《RNA-seq方法和应用》这一集。自己去看一下。
那么,我们来说GO分析。
这张图是GO分析的结果。
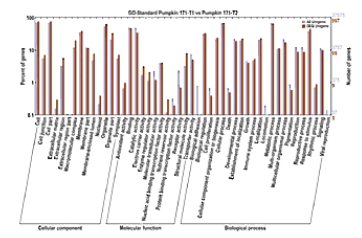
这是根据突变的点在肿瘤中的富集的情况做的分析。
分成“细胞组件”、“分子功能”、和“生物过程”3个大类,进行展示。
柱子越高,则表示这个亚类当中突变越多。
这是有向无环图,它把突变进一步一步富集到更精细的小概念当中进行展示。
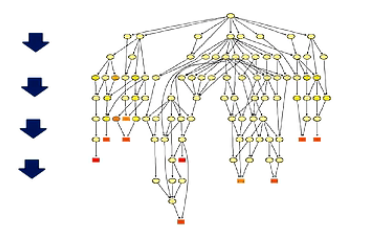
这个图中,是越向下,它的功能就划分得越精细。同时颜色越深的块块,则表示突变在
这个小概念中富集程度越高。
这是Pathway的KEGG富集分析。它吶,可以帮助我们看到哪些通路发生了显著的变化。

更深一步的分解分析,则让我们深入地看到突变的基因在整个通路中所处的节点。
让我们更好地探索突变和病变之间的关系,我们要说明:在外显子测序的数据分析当中,对基因组的结构变异是不敏感的。
不敏感的原因是外显子测序,只测了基因组上很小一部分区域,这个比例小到了只有1~2%
,所以当结构变异的断点,不落在外显子区域的时侯呐,外显子测序是看不到这些断点的,所以我们说:外显子测序对基因组的结构,变异--SV(Structure Variation)呐,是不敏感的。

对CNV不敏感
外显子测序对拷贝数变异(CNV,copy numbervariation),不是很敏感。不敏感的原因呐,是因为杂交捕获过程啊,是一个含了很高偶然性的过程。
也就是说,一个外显子片段上,它有多少个reads(序列)被捕获下来,样本和样本之间是有很大差异的。
或者说,它的覆盖度,本来就是忽高忽低的,因为有这种忽高忽低呐。
这就导致:一个外显子上测到的Reads数变高,或者变低。
我们很难判断,是因为这种偶然性呐,还是因为拷贝数发生了变异。所以,外显子测序,对于小片段的拷贝数变异,它本身是不敏感的。
但是,如果基因组上发生了大片段的拷贝数变异。比如说,长度在5M(5M base pair)以上的
片段发生了拷贝数变异。那么外显子测序呐,是可以发现的,这是因为这样长的片段当中呐,一般含有多个外显子。当多个外显子的测序Reads数都发生了改变,那么,它就会有统计上的显著性。通过这种统计上的显著性,我们可以来判断:基因组上的确发生了拷贝数变异。

因为外显子组测序对结构变异和拷贝数变异不敏感,所以,在实际的肿瘤基因测序中呐,科学家往往是这样做的:用全基因测序来找到肿瘤样本中的结构,变异(SV)和拷贝数变异(CNV),再用来外显子组测序来找肿瘤样本中的、低频的SNP和Indel体细胞突变。
捕获Panel测序
今天,我们在讲外显子组测序的同时呐。我们就顺带说一下针对某些疾病所设计的捕获Panel测序,所谓Panel,往往是指对若干个基因设计一个捕获试剂盒。
诊断公司为诊断特定的疾病,设计了许多特定的、针对性的Panel。
例如著名的肿瘤诊断公司Foundation Medicine就设计了“Foundation One”这个Panel。
它(Foundation One)是针对实体瘤的一个Panel,这个Panel包含了315个经常发生突变
的肿瘤相关基因。还包含了28个经常发生重排的基因。
这一类的Panel,它的建库、捕获、和测序原理,与外显子组测序是完全一样的。
但是因为它所选择的基因数远少于外显子,所以就可以用较少的测序量得到非常深的测序深度。
同时因为测序数据量较小,所以数据分析的难度也会小许多,耗时也会更短。这对于临床诊断所需的快速响应呐,是有十分多的好处。
3. small RNA-seq
今天要给大家介绍的是:small RNA-seq,也就是“小RNA的测序”。
那么,小RNA呢,包括了micro RNA/tRNA/piRNA等一系列的、片段比较短的RNA。其中,micro RNA因为其基因数量众多,同时,表达量变化丰富,是近10年来的一个研究重点,我们今天分2部分来介绍samll RNA测序。
第1部分是介绍small RNA的建库测序方法。
第2部分是介绍small RNA的生物信息学分析。
建库方法
那么,我们先说第1部分,small RNA建库。

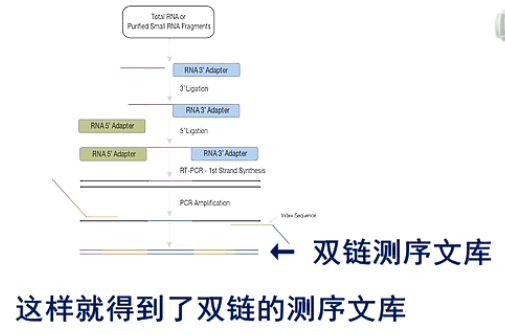
这张图是small RNA建库的流程图。
在small RNA的结构上,都是5’端有一个磷酸基团,在3’端有一个羟基基团。


在建库过程中,先在它的3'端连上一个3'端专用的接头。然后,再在5'端连上一个5'端专用接头。

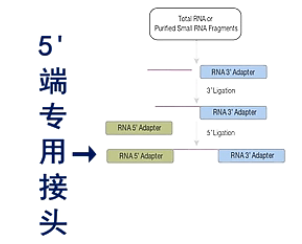
然后进行逆转录,得到第一链的cDNA。

接着再进行PCR扩增
这样就得到了双链的测序文库。
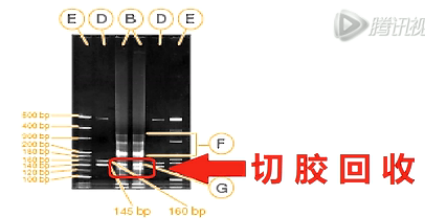
这张图,就是建好的small RNA文库。
用Agilent Bioanalyzer 2100进行电泳,得到的电泳图。

如图所示,扩增之后得到的small RNA的文库。
在整个的扩增混合物中,只占很小的一个比例。
所以,一般情况下,这个文库还要经过进一步的电泳胶分离。切胶回收,才能得到比较纯的、我们要的small RNA文库。
纯化好的文库,再用Agilent Bioanalyzer 2100进行电泳。
我们就可以看到比较纯粹的small RNA文库了。

目前用illumina Truseq small RNA建库试剂盒。
对组织中抽提到的总RNA进行small RNA建库。
一般一个反应需要1微克的总RNA。
同时small RNA建库,对(总)RNA的质量也会有一定的要求。
一般是要求总RNA的RIN值大于等于8.0。
关于RIN值(RNA Integraty number,RNA完整度值)的意义,如果观众有不清楚的,又想要了解的,可以在优酷视频当中找一下【陈巍学基因】《视频7:RNA-seq》,里面有专门的介绍。
生物信息分析
接下来,我们介绍第2部分:small RNA的生物信息分析。
small RNA生物信息分析的第1步,是把测序的序列进行过滤。
也就是把引物二聚体、和含有多个N的这些序列去掉。
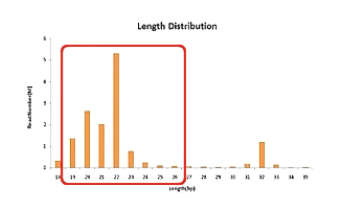
然后,就是统计各种长度的small RNA各有多少条。
一般情况下,人源组织所测到的small RNA会在22BP左右有一个主峰。这个主峰就是micro RNA,同时,30BP左右又会有一个副峰,这个峰,主要是piRNA。

接下来,就是把small RNA,比对到参考基因组上。
在参考基因组上比对好之后,就可以把这些序列和已知的small RNA数据库进行比对了。
比较有名的small RNA数据库是miRBase,这个数据库目前这个数据库已经收录了2000多条人源的micro RNA基因。
在对人源样本的测序过程当中,大家最关心的主要是micro RNA和piRNA,这2种small RNA。那么在测序过程当中,实际上还会测到rRNA的碎片和tRNA的序列。
因为rRNA和tRNA在人的基因组中是十分保守的,所以一般不是我们关注的重点。
对表达量的分析
对已知small RNA的分析,主要是对表达量的分析。
small RNA的表达量,一般用TPM来衡量。TPM是Transcripts Per Million reads的
缩写。
也就是1百万条测到的序列当中,某个目标small RNA的序列条数。
TPM的密度分布图,能整体展示样本的small RNA基因表达情况。
图中,横轴是一个small RNA基因的表达量。越向右呢,则这个基因的表达量越高。纵轴是有特定表达量的基因数量,越向上,则基因数目越多。
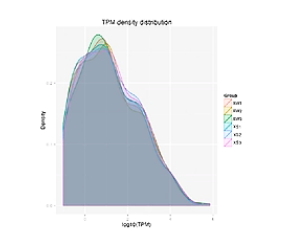
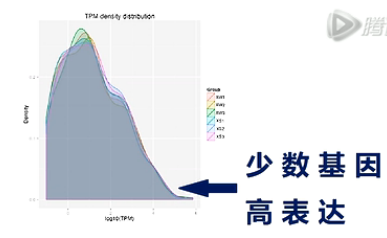
从这张图上可以看出,少量的基因有高表达,大多数基因的表达量,还是相对偏低的。
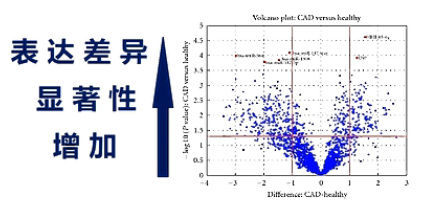
用火山图,则可以整体地观察两个样本之间的表达差异。
火山图的横座标,是某个small RNA基因的表达的增减。从0向右,则表达量上升,从0向左,则表达量下降。纵轴则是表达量差异的显著性,越向上,则差异越显著。
一张火山图,可以让我们轻松地观察2个,样本之间,small RNA的表达差异。
聚类分析,则可以帮助我们直观地观察,一批样本当中,那些样本有共同的表达特征。又有哪些small RNA基因有相似、相近的表达量。
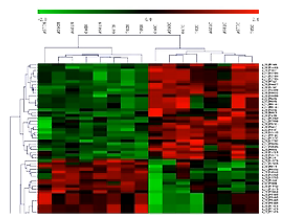
如这张图中所展示,样本经过聚类分析,明显地可以看出,其small RNA的表达谱,呈现2种表达情况,上绿下红的样本呢,自然地被分到了一组,上红下绿的样本呢,就会被自然地分到另外一组。
通过聚类分析,我们可以观察到样本内在的共同特征。
在人类细胞中micro RNA主要是通过和mRNA结合,来阻止mRNA翻译成蛋白,从而起到抑制靶基因表达的作用。目前,只有少数的micro RNA和靶基因mRNA的对应关系是经过了实验验证的。大多数还是通过序列互补、结合热稳定性等预测性手段来预测的。
所以,这些关系不是很精确的。虽然这种预测不是很精确,但是它能为我们的科研提示有用的研究目标。
靶基因 GO 和 Pathway 分析
接下来,我们说一下micro RNA侯选靶基因的GO分析、和KEGG Pathway(通路)分析。
GO分析和KEGG Pathway分析是非常常用的生物信息学分析手段。
想要了解这2种分析的基本情况的同学,可以在优酷中找一下【陈巍学基因】《视频7:RNA-seq》去看一下。
通过GO分析,表达差异被富集到分类的GO的子项目当中,通过这个图,可以看到“生物过程”、“分子功能”、和“细胞组件”的哪些环节出现了明显的差异。柱子越高,则表示差异越明显。
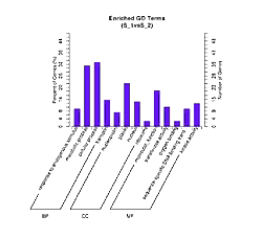
有向无环图,是进一步把差异一步一步地富集到更精细的小概念当中进行展示。
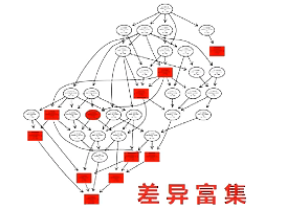
在这个图当中,越向下,功能就越是细分。同时,颜色越深的方块呢,则表示差异在这个小概念当中,富集程度越高。通过对表达差异的micro RNA和它对应的靶基因进行KEGG分析,
可以把可能被影响到的通路进行富集分析。
这个图,就是KEGG分析的结果。在此图中,KEGG富集的程度,通过富集因子、Qvalue、和富集到此通路上的基因个数,来进行衡量。
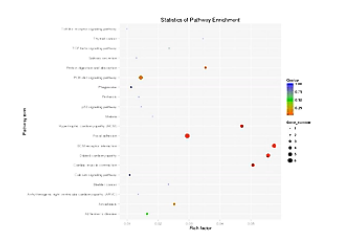
点的面积越大,则富集的基因数越多,富集因子越大,则表示富集的程度越大。
接下来这个通路图,是对某个特定通路的进一步的细化分析。
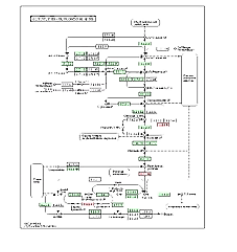
它可以让我们看到,在一个整体的通路中,具体是哪个、或哪几个节点会有显著的差异。
寻找新的 micro RNA 基因
寻找到新的micro RNA基因。一般是测序测到新的、有发夹结构的microRNA前体的序列,同时测到对应的成熟的micro RNA序列,并且在基因组上又找到了对应的基因序列,这样,大体上就判断(可能是)找到了一个新的micro RNA基因了。
以上,我们说的都是组织中的small RNA的测序和分析。
血浆 micro RNA 测序
随着技术的持续进步,目前用血清、或者血浆中的micro RNA来诊断疾病,成为诊断医学十分关注的一个研究方向。这是因为:
1) 血清当中有大量的、种类丰富的micro RNA。并且这些micro RNA可以相对稳定地存在
2) 同时我们已经知道micro RNA参与多种基因的表达调控
3) 血液又是我们最容易获得的诊断样本之一
4) 而且,目前血清、或者血浆中的micro RNA已经可以被方便地测到
所以,许多学者都在研究血清micro RNA,以期望从中找到新的诊断Biomarker。
目前,做一个血清micro RNA测序,大约只需要0.5毫升左右的血清、或者血浆。
也就是相当于1毫升的原血就够了。
用于micro RNA测序用的血清、或血浆,可以用3倍体积的Trizol LS来进行保存。也就是说,0.5毫升的血清,加上1.5毫升的Trizol LS。混合均匀之后呢,-20℃、或-80℃保存。然后,通过干冰运输,交给专业的测序公司,就可以进行测序、分析了。
4. 单细胞DNA测序
今天,和大家谈一下单细胞测序。
自从二代测序技术出现,把一次实验测许多条DNA序列的这个难题解决之后,一次把一个人的全基因组给测出来,最极限的情况,就是样本量就是少到一个细胞,就要测出整个基因组的序列信息。
三个难题
要实现从一个细胞样本测出全基因组的DNA序列,至少要克服以下3个难题:
第1个,就是如何实现均匀扩增,也就是说,用传统的随机引物PCR的方法来扩增。那么不同扩增片段的扩增效率多少会有一些差异,这些扩增效率的差异会随着扩增循环数的增加,呈现出指数放大的效果。其结果就是会发生严重的覆盖不均一,极少数区段的DNA被大量扩增,测序后它深度非常深,但在大多数区段只有很低的覆盖,甚至没有覆盖。那么我们就无法有效地判断那些低扩增效率区段的基因序列的情况。
那么它的第2个难题,就是 全基因组覆盖问题。常规的、用大量DNA进行建库的方法,因为打断、补平、加A、加接头等一长串的操作,每一步都会有DNA片段的损失。结果就是初始DNA中很大一部分会被浪费掉,而没有形成有效的文库分子。
在单细胞测序中,丢失大部分的起始DNA,是不可接受的。单细胞测序要求几乎所有的原始基因组片段都得到扩增,并且在后续的测序过程中被测序测到。这就要求几乎所有的片段,都会被得到扩增,而不只是少数片段得到有效扩增。
第3个难题,是这种方法要有较高的扩增效率。建好的文库,在HiSeq测序仪上机的时侯,大约每上机2万个文库分子,只有1个文库分子,是能够在测序的Flowcell表面生成簇,并且被测序测到的,剩下的大多数文库分子,在上机的时侯是被水冲走的。所以,单细胞基因组扩增的方法,还要有较高的扩增效率。至少要有上万倍到几十万倍的扩增效率,才能保证在全基因组测序的时侯,大部分的片段都被测序测到。
两种方法
为了解决上述的难题,科学家想了许多的办法。
到目前为止,大家比较认可的方法有两种:
第一种是MALBAC方法。
第二种是MDA方法。
MALBAC方法
我们先来说这个MALBAC方法。它的全称是:MultipleAnnealing and Looping-Based Amplification Cycles。是谢晓亮教授发明的方法,
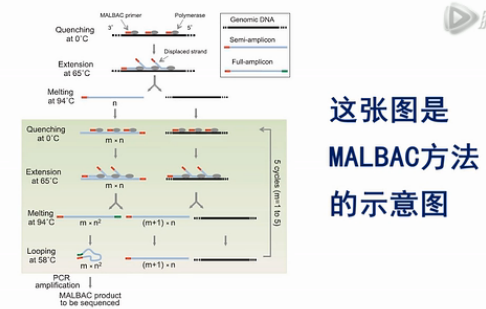
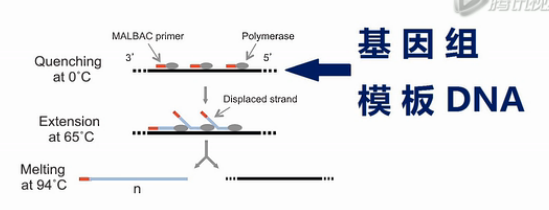

这张图是MALBAC方法的示意图。这个黑色的线条,就是基因组模板DNA,这些红颜色的线条就是扩增引物,扩增引物的5’端有27个碱基的通用序列,这些通用序列会作为未来的PCR通用扩增引物的结合序列。扩增引物的3’端有8个随机序列的碱基,这8个碱基可以随机地杂交到基因组DNA的互补序列上。
这些灰色的椭园是Phi 29 DNA聚合酶,Phi 29 DNA聚合酶有一个特点,它不仅可以生成新的DNA链,它还能把之前已经合成好的DNA链给解链开。

再形成自己的新链,这个特点能够把每个循环所能合成的DNA新链的数量提高几倍、甚至几十倍、上百倍。
接下来,就是做5个MALBAC循环,请注意,这里每个循环的最后一步是58度退火。我们后面要详细解释这一步58度退火的作用。
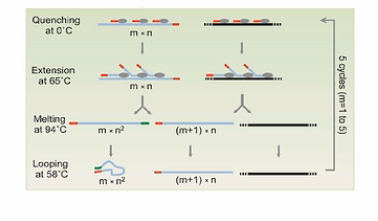
第一个循环下来,得到的是一批5’端有通用扩增序列的DNA片段。
在第二个循环完成后,所产生的扩增产物中,大部分是5’端有通用序列。而3’端,有与通用序列互补的序列的这些片段。
图中的这4个步骤,一共重复5次,这样做的巧妙之处,就是要解决我们刚才所说的3个难题。
第一、是要均匀扩增
第二、是要全基因组覆盖
第三、是要有高的扩增效率
那么,我们先来看这个线性扩增。
刚才,这个MALBAC方法的巧妙之处,就是在每个循环的最后,加了一步58度退火,这一退火过程,它让完整扩增的产物,它的两端发生链内杂交。这样,3’端的序列就不能与新的、游离的引物发生杂交。这也就不会引新的、发起始于3’端的扩增,这样,就避免了完整扩整的产物的自我指数扩增。

现在,还是8个随机序列的引物在模板上随机地找结合位置,所有的位点都有一样的机会被扩增。
那么,这样实际得到的产物分3种:
第1种,就是m* n 个“完整扩增产物”,这是最主要的产物。这里“m”就是循环的次数, “n”是一个循环中,有多少个扩增,引物可以粘到一个模板上。
第2种扩增产物,就是(m+1)* n个“半扩增产物”,第3种DNA,就是那个原始的DNA模板,这里完整产物的数量是“m*n ”,也就是说,扩增产物(的数量)与扩增的循环次数“m”成正比,而不是与m的平方成正比。更不是与2 的M次方成正比。
这也就是达到了,我们想要的“线性扩增”的目的。也就是说扩增产物(的数量)与扩增的次数成线性关系。这就达成了我们单细胞测序当中第1个要求“线性扩增”。
第2个要解决的难题,就是“全基因组覆盖”
这里,是利用Phi 29聚合酶的能一次在模板上聚合出多个新链的功能来达到这个目的。
在5轮的扩增之后,每个模板都会有5*n^2个扩增片段。这样,就可以保证建库时大多数的
基因组区域可以被建成文库,最后,可以被(测序)测到。
第3个要解决的问题“高效率扩增”。还是利用了这个Phi 29酶的一次得到多个扩增片段的这个效果,来达成的。
上面所说的,就是MALBAC单细胞扩增技术的基本原理、和它的巧妙之处。
MDA方法
目前市场上还有一种单细胞的扩增技术,叫MDA扩增技术。它的全称是MultipleDisplacement Amplification。
MDA方法的技术核心是用Phi 29 DNA聚合酶来进行直接的扩增。
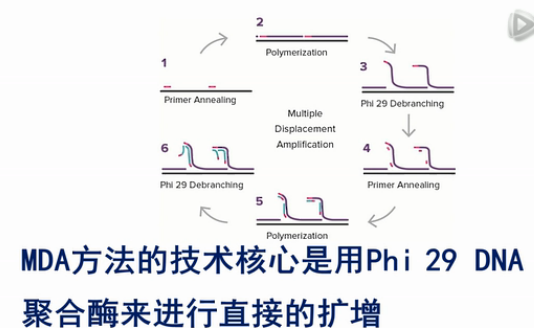
Phi 29酶的特点是,它可以把双链DNA进行解链,然后,在常温条件下,就把原始模板进行大量扩增。
两种方法的比较
把MDA和MALBAC两种方法进行比较
MDA的优势在于,它的扩增效率更高,并且,实验方法更简单。
MALBAC方法的特点,在于它的扩增均一性更好。但是,它得到的扩增DNA量相对较少,或者说,它的扩增效率相对比较低。
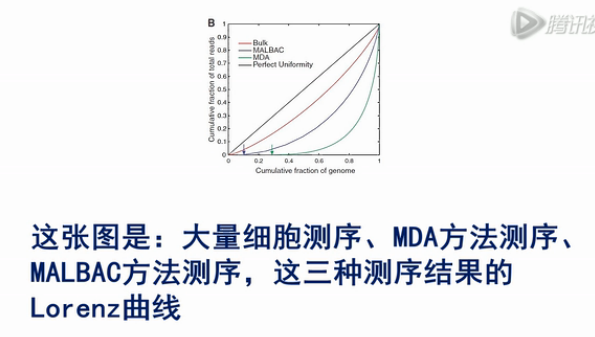
这张图是:大量细胞测序、MDA方法测序、MALBAC方法测序,这三种测序结果的Lorenz曲线。
Lorenz曲线是越接近于对角线,则覆盖越均一,从图中,我们可以看出大量细胞测序,它的均一性是最好的。
用MALBAC方法测序,它的均一性比大量细胞测序的均一性要差一些,但是要比MDA的方法的均一度要好。
这张图是用三种方法来测肿瘤细胞中的拷贝数变异。其中横轴是染色体的序列,纵轴是测序的覆盖深度,可以明显地看到,在大量细胞测序的结果中,可以非常直观地看到拷贝数变异的情况。
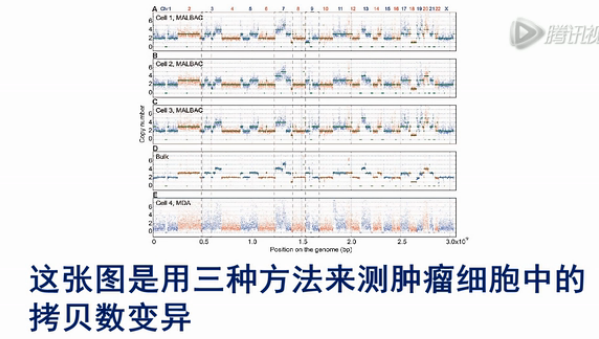
而用MALBAC的方法,也还是能够比较清楚地看到拷贝数变异。但是,它没有大量细胞测序的结果那么清楚。
而用MDA的方法来看拷贝数变异,则不是那么容易看清楚。
临床应用
单细胞测序,有着广泛的应用前景。目前最主要2个应用:1个是在胚胎植入前进行基因拷贝数变异检测。第2个,是进行肿瘤的染色体变异研究。
在这里我们介绍一下,单细胞测序在胚胎植入前检测中的应用,在有习惯性流产的夫妇当中,最常见的病因就是染色体的平衡易位,所谓染色体平衡易位,也就是A染色体,的一段移到了B染色体上。
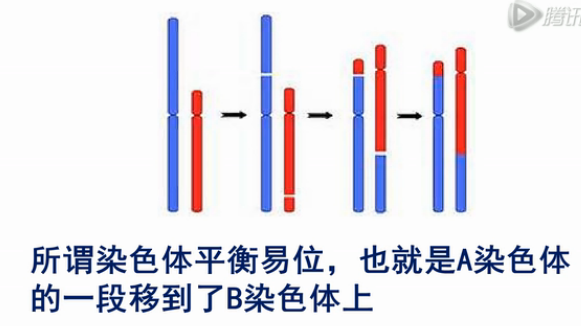

如果夫妻一方有染色体平衡易位,那么这对夫妇的受精卵中,每4个受精卵,可能只有1个是正常的。剩下3个(不正常的受精卵),很可能会流产。
要把这一个正常的受精卵挑出来,目前,最有效的解决手段是做受精卵植入前检测。
那么具体的操作方法,就是先做人工受精。
在受精卵发育到8个细胞的时侯,通过显微操作,抓一个细胞出来进行测序。
在这个测序过程当中,就要用到MDA方法或MALBAC方法进行扩增、建库、测序。
然后测序完成之后,挑出那个好的受精卵,植回到母亲的子宫中去。长成一个正常的新生儿。
这个,就是受精卵植入前基因检测。
这项技术,是对生殖健康有很大帮助的一项新技术。
5. 单细胞mRNA测序
今天,想和大家谈一下单细胞mRNA测序技术。
单细胞mRNA测序一直是科学家关注的一个热点。目前市场主要有2种建库方法,分别是Clontech公司推出的SMART法,和EpiCentre公司推出的TargetAmp方法。
要实现单细胞mRNA测序,需要解决2个难题。
第一个难题:PCR偏差
第一个难题就是一个人类细胞当中,它的总RNA量大约只有10pg左右(1pg=10^-12g),中的mRNA的量大约只有0.2个pg。要把那么少的mRNA转变成约零点几个μg(1μg=10^-6g)以上的核酸文库,这意味着核酸的扩增量要达到几百万倍以上。
如何能在这个核酸扩增过程当中不引入太多的PCR偏差,就一直是个大问题。
所谓PCR偏差,就是在PCR扩增过程当中,某些片段被大量扩增,而大部分片段被扩增的量很少,甚至根本就没有被扩增。结果就导致高通量测序,只能测到这所有样本当中很少一部分的片段序列。
PCR偏差会随着PCR循环的次数的增多而指数放大。那么,在这种情况下,一方面要把核酸扩增几百万倍,甚至更多的倍数;另一方面,又想得到均一覆盖的文库,这就是单细胞mRNA建库当中,所要解决的第一个大难题。
第二个难题:去除核糖体RNA
第二个难题是如何能尽可能高效地得到“mRNA”的文库,而不是含了大量“rRNA”序列的文库。因为rRNA在总RNA当中占了95%,甚至更高的比例,而mRNA在总RNA当中只占2~3%的比例。如果不加区分地进行逆转录,再扩增、建库很可能测序得到的绝大部分序列都是rRNA的序列。
但是 rRNA序列不能给我们带来有效的生物信息,它是无用的。而只有测到mRNA的序列,才是我们想要的信息,这样,如何能够选择性地把mRNA转化成测序文库,并且避免把rRNA带到测序文库中来,这就是单细胞mRNA测序当中,要解决的第二个大难题。
接下来,我们就来介绍SMART方法和TargetAmp方法,是分别如何解决上述2个大难题的。
SMART方法
我们先来介绍Clontech公司推出的SMART方法。
SMART方法的全称是Switching Mechanism at 5’ End of RNA Template。
这张图就是SMART方法的原理图。
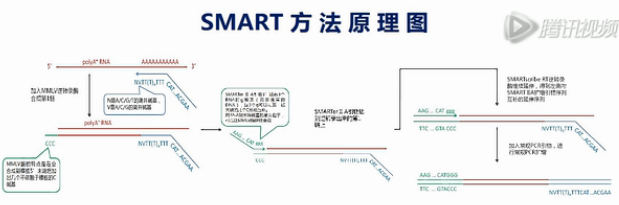
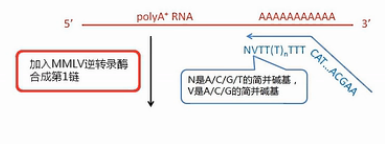
SMART方法最核心的技术,就是设计了2个特殊的引物。再配合用MMLV逆转录酶进行逆转录。

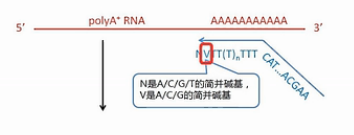
我们先来看这个逆转录的起始引物。它哪,先是一段通用序列,未来这个通用序列会用作PCR扩增的引物识别序列,中间是一长串的T,这些T是专门识别mRNA的3’末端的Poly(A)尾巴序列。它会和这些Poly(A)尾巴互补结合,引物的最末端有一个定位的结构,这个定位的结构,就是在它的3’末端的倒数第2个碱基是一个非T的简并碱基。
图中用V来表示这个碱基。V碱基就是或A、或C、或G,但是非T的这样一个(简并)碱基。
最后1个碱基则是简并碱基N,也就是A/C/G/T都有可能。
引物的这个末端结构,就是让它正好结合在mRNA的3’端连到Poly(A)尾巴的这个连接处,而不会结合到mRNA的别的地方。这样就保证了逆转录的起始位置正好是mRNA的3’端的序列终止位置。
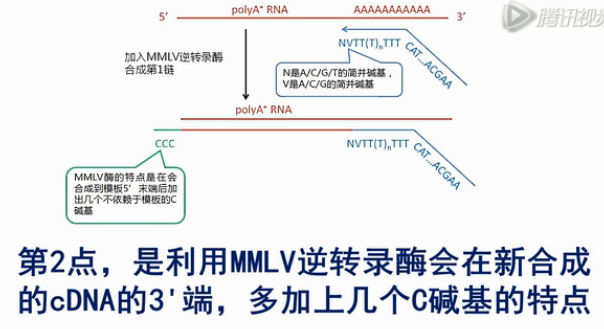
我们再来看这个MMLV逆转录酶,这个酶有个特点,就是它在转录到mRNA的5’端末端的时侯,会在新合成的cDNA的3’末端,多加出几个C碱基来。

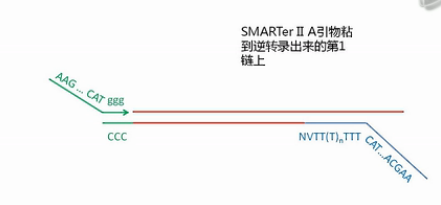
再接下来,这个上游引物会发挥它的作用。这个上游引物,它有一个特点,它的3’端是3个非脱氧的G碱基,也就是核糖核酸的、RNA的G碱基,而不是DNA的G碱基,这个引物可以与刚才新合成的cDNA的3’端的那几个C碱基发生互补杂交,然后引导这个MMLV酶再次发挥聚合作用,以刚才那条新合成的cDNA为模板,复制的结果,就是得到双链的cDNA。
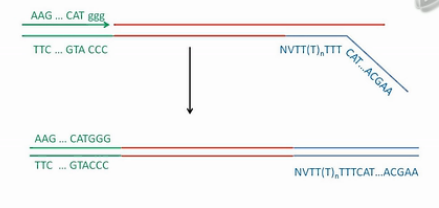
这个双链cDNA,两端都已经接好了我们人工设计的PCR引物序列,然后,就加入常规的PCR引物,进行常规的PCR扩增,常规PCR扩增,得到大量DNA。然后可以象常规的DNA建库那样,超声打断、建库、上机测序了。
我们回顾一下这个过程,可以看到这个方法的3个巧妙点。
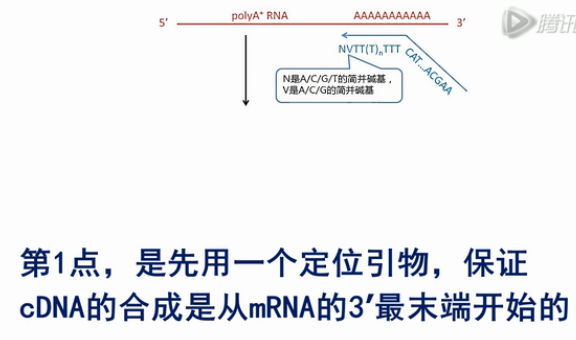
第1点,是先用一个定位引物,保证cDNA的合成是从mRNA的3’最末端开始的,同时让合成出的cDNA在下游连上了一个通用PCR序列。
第2点,是利用MMLV逆转录酶会在新合成的cDNA的3'端,多加上几个C碱基的特点,再用有3个G碱基的上游引物进行第二链的合成。这就保证只有完整的第一链cDNA也就是那些带多个“C”的cDNA(第一链)才能被合成出cDNA的第二链,这也就保证了双链cDNA是全长的cDNA。
第3点,就是要保证PCR扩增的效率的一致性那我们知道,PCR扩增效率的最主要的影响因素是引物的序列,现在因为cDNA的5’端和3’端的都分别引入了统一的引物序,所以,这就去除了因为引物序列的不同。而引起PCR效率不同的,这个最主要的偏差因素。这也就在较大程度上保证了PCR扩增效率的一致性,减少了PCR偏差。
经过实验发现
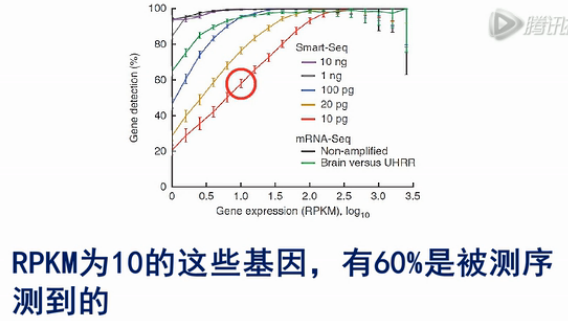
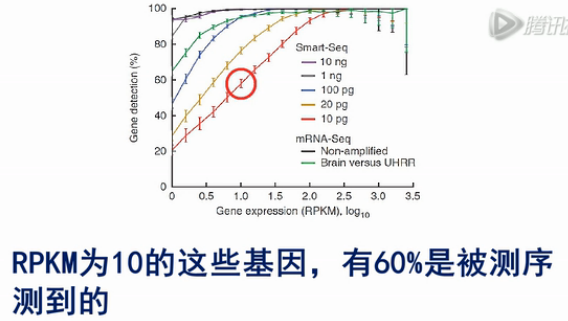
用SMART方法,对于1个细胞,也就是10pg总RNA的RNA进行建库测序,RPKM为10的这些基因,有60%是被测序测到的;对RPKM为100的基因,有90%可以被测序测到。而且被测到的几率,波动很小。这说明SMART方法是一个有效的单细胞mRNA测序方法。
TargetAmp方法
接下来,我们介绍第2个单细胞mRNA建库的方法--TargetAmp方法
这个方法是由Illumina公司旗下的EpiCentre公司开发的。
这个就是TargetAmp的原理图。
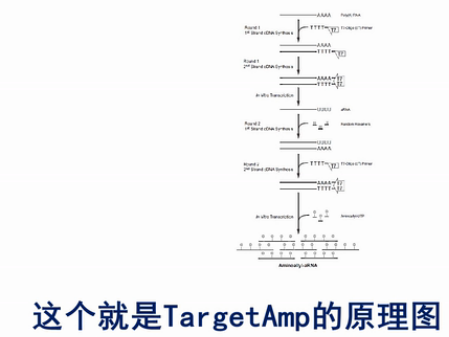
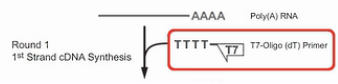
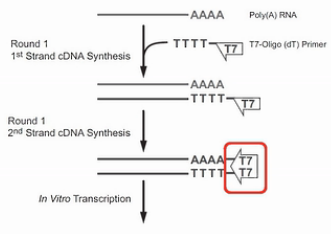
首先是用T7-Oligo(dT)的引物进行cDNA合成。这个引物,在5’端有设计了一个T7启动子序列。3’端是多个的T碱基,这一串T碱基可与mRNA的poly(A)尾巴相结合,作为逆转录的起始引物,经过逆转录,得到第一链的cDNA。同时这条cDNA链上,被引入了一个T7启动子。

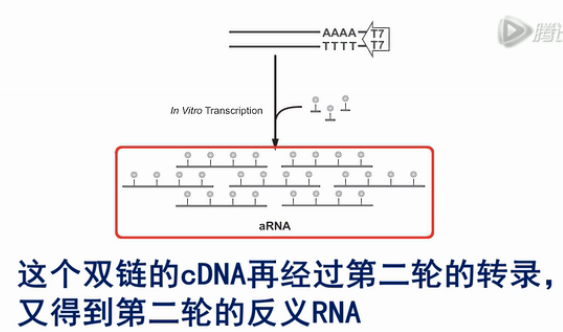
然后用RNase H酶把cDNA:RNA双链产物中的这个RNA链消化掉,接着再合成出第二条cDNA链来,这个双链的cDNA就可以作为转录的模板。利用链上的T7启动子,转录出大量的反义RNA来(antisense-RNA,aRNA)。

接着,将这些反义RNA进行纯化。再用随机引物进行逆转录,得到第二轮的cDNA,接着,再用T7-Oligo(dT)这个引物,粘到第二轮的cDNA的Poly(A)尾巴上,再合成出双链DNA来。


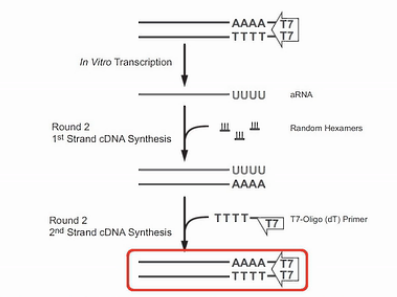
这个双链的cDNA再经过第二轮的转录,又得到第二轮的反义RNA,这些第二轮的反义RNA的量,足可以达到微克级。再经过一轮逆转录,就可以得到几个微克的cDNA。那么几个微克的cDNA,就足以进行建库、测序之用了。
我们来看TargetAmp方法的巧妙之处:
它不是用PCR来扩增核酸,而是用转录的方法来增加核酸的量。因为扩增那么多(倍)的核酸,如果用PCR,要用几十个循环,那么PCR不同的扩增子的扩增效率,即使一开始是很小的差异,也会在几十个循环中,被指数放大,变成一个很大的差异。
那么TargetAmp方法用转录的办法,而且统一都用T7这个统一的启动子,它转录的启始效率,大体上就保持了一致。
它的每一轮转录,都把核酸的量扩大几千倍。经过这样两轮的扩增,就把核酸的量扩大了几百万倍。这样,一方面它得到了高达几微克的核酸。足够用于建库,同时又避免了PCR过程,也就避免了PCR扩增偏差
单细胞mRNA测序方法,在循环肿瘤细胞研究、胚胎发育研究、和神经活动研究方面,有着广泛的应用。
随着高通量测序的费用不断地降低,它正变成科研中越来越普及的研究手段。相信有更多单细胞mRNA建库方法,和更新的技术应用会不断地被开发出来
6. 甲基化测序
本期节目,要给大家介绍一下DNA的甲基化和羟甲基化的高通量测序。
DNA的甲基化是在DNA的序列不变的条件下,在其中某些碱基上加上甲基的这样一个过程。
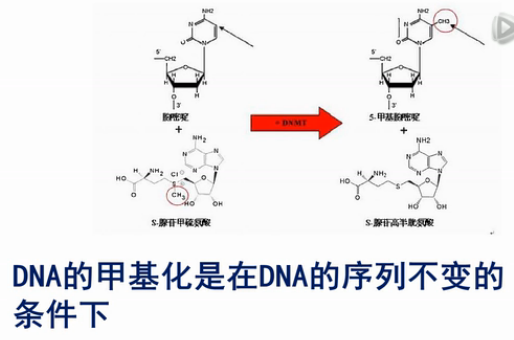
DNA甲基化的结果,一般是使甲基化位点的下游的基因表达量变少。
化学反应
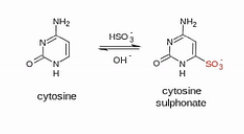
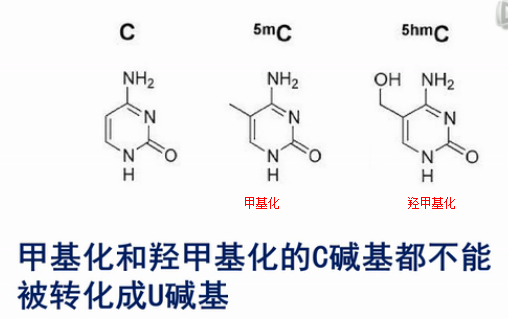
这个(甲基化)分析方法当中的核心化学反应,是用亚硫酸氢盐来处理DNA。DNA当中,没有甲基化或羟甲基化的C碱基,就会被转化成U碱基。
我们来看这个转化的过程,在弱酸性条件下,亚硫酸氢根会结合到没有甲基化的C碱基的6位。而甲基化了的C碱基不会和亚硫酸氢根发生这个反应的。
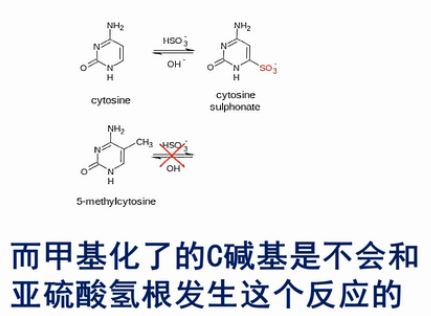
然后,用碱来处理。结合了亚硫酸氢根的非甲基化的C,就被脱氨基,并且脱亚硫酸根。然后,就被转化成U碱基。
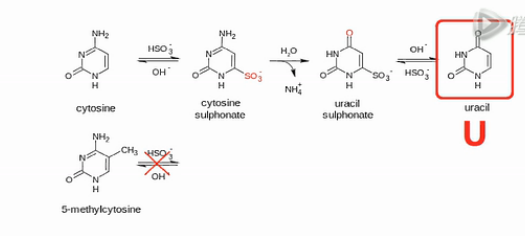
那么,甲基化或者羟甲基化的C碱基,因为之前没有和亚硫酸氢根起反应,所以现在用碱来处理,它也不会发生脱氨基反应。所以,它还保持了是“C”。
用亚硫酸氢盐来处理DNA,可以让99%左右的非甲基化的C碱基变成U。
也就是说这种方法的的转化效率非常高,转化效率达到了99%。
它的优点,就可以让我们接下来通过高通量测序的方法,可以精确地看到单个碱基的甲基化的水平。
经过亚硫酸氢盐转化过的DNA,再经过PCR,PCR新合成出来的链,U碱基的位置,就会被替换成了“T”。那么在接下来的测序过程中,测到的也是T碱基。
而甲基化的C,因为没有被亚硫酸氢盐所转化,所以,在接下来的测序过程中,被测到的,还是“C”碱基。
这样,通过测序,看一个位置是“C”,还是“T”。如果它保持是“C”,就说明这个位置是被甲基化、或者羟甲基化了。如果测到的是“T”,就说明这个位置是没有被甲基化、或者羟甲基化。
建库方法
接下来,我们谈一下甲基化的建库过程。
先说第一种,用Illumina公司的Truseq DNA建库方法,来做甲基化测序。
因为Illumina Truseq DNA建库试剂盒当中,它所提供的接头,那么这个接头上的C碱基都是已经经过甲基化修饰了。
所以,用这些接头做出来的文库,在用亚硫酸氢盐做转化的过程当中,它的(接头上的)C还是保持是C ,不会被转成U。
带了这些接头的文库分子,就可以和测序芯片上的草皮DNA发生互补杂交。并且进一步发生桥式PCR反应。生成测序用的DNA的簇(Cluster)。
但是,这个方法有一个缺点,就是在用亚硫酸氢盐处理DNA文库的时侯,90%以上的DNA链会断掉。这样,已经建好的文库,其中90%分子会被破坏掉。也就是说文库的丰富度就会损失90%以上。
那么,相应的它有它的好处,它的好处就是,在这个建库过程当中用的PCR循环数较少。所以它PCR扩增效率不同,所引起的文库不均一程度也就较低。也就是我们通常所说的PCR bias较少。
接下来,再说第二种建库方法。
为了解决文库丰富度受到损失的这个问题,EpiCentre公司开发了EpiGnome方法,方法的操作过程如图。
第1步,亚硫酸氢盐先处理DNA,把未甲基化的C都转变成U。

第2步,把带标签1的随机引物加入,进行第一次的复制。得到第1条的复制链。

第3步,是消化掉过量的引物。
第4步,是加入带末端终止碱基、并带标签2的随机引物。这个引物的作用是让第1复制链延伸,并且加上标签2。
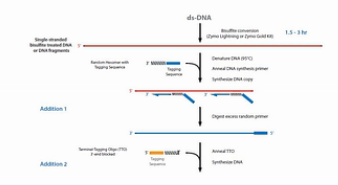
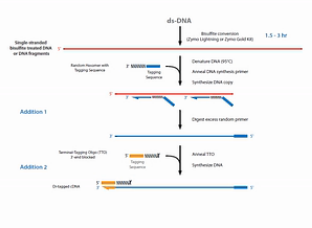
第5步是加入建库的PCR引物,进行PCR。通过PCR,把Index序列和成簇引物序列加入到链的两侧。得到真正的文库。
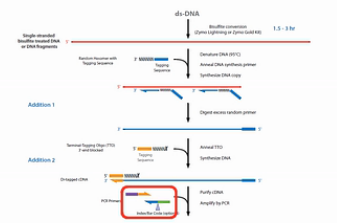
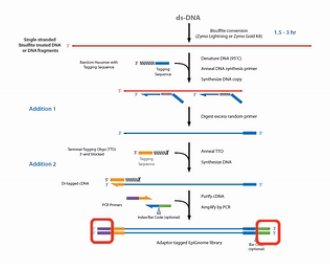
这个方法的优点是,把亚硫酸氢盐处理的过程,放在了建库之前。这样建成的库的丰富程度会比较高。但是这个方法也有缺点,缺点就是要做较多的PCR循环,那么有了比较多的PCR循环之后,PCR产物的扩增均一性是不太好的。也就是说PCR bias会比较大。
上述两种方法,各有优缺点。
HiSeq2000/2500测甲基化文库的问题、和解决方案
在Illumina的HiSeq 2000或者2500平台上进行测序,如果文库是碱基平衡的文库,也就是说,每个特环当中,A/C/G/T四种碱基的比例,各占25%左右的话,测序仪对碱基的判读会比较好。
但是如果缺少了一种或者几种碱基,测序仪对碱基的判读就会出问题。测序得到的数据质量就会下降。并且效的数据产量也会降低。
关于文库碱基平衡度影响数据质量和产量的原因,在【陈巍学基因】第二期的视频,HiSeq义器的工作原理这一集中有介绍。大家可以在优酷当中找一下,并且看一下。
因为甲基化文库中经过亚硫酸氢盐处理,绝大多数的C都变成了T。所以,这个文库中是严重地缺少C碱基的,也就是四种碱基的比例是严重不平衡的。这样在用HiSeq 2000或2500测序仪来测甲基化文库的过程当中,文库测序得到的数据质理就较差。并且经过PF过滤得到的有效的数据产量也会较低。
为了弥补甲基化文库的碱基不平衡性,一般情况下,在上机过程当中,是掺入大比例的基因组文库,或者PhiX文库,来补充比较多的C碱基,一般会掺30%的PhiX文库、或者基因组文库。
在掺入30%的PhiX文库的条件下,一条HiSeq 2000 V3 PE100的Lane,大概可以得到20G 左右的甲基化文库数据。
也就是说,在HiSeq 2000或者2500平台上,甲基化文库的测序数据产量,一直都不是很高。质量也比较低。
羟甲基化测序
接下来,我们说一下区分“羟”甲基化和甲基化的测序方法。
在用单纯的亚硫酸氢盐法来测的过程当中,甲基化和差甲化的C碱基都不能被转化成U碱基,所以单纯的亚硫酸氢盐法是无法区分甲基化或羟甲基化的C碱基的。
为了区分甲基化和羟甲基化,科学家想出了两种办法。
第一种办法,是通过高钌酸钾(KRuO4)来氧化羟甲基化的C。羟甲基化的C可以被转化成甲酰化的C碱基,而甲酰化的C碱基,是可以被亚硫酸氢盐转化成U的。
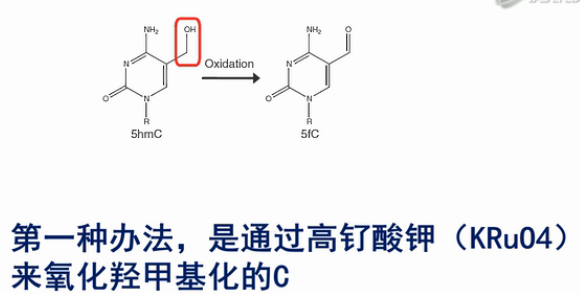
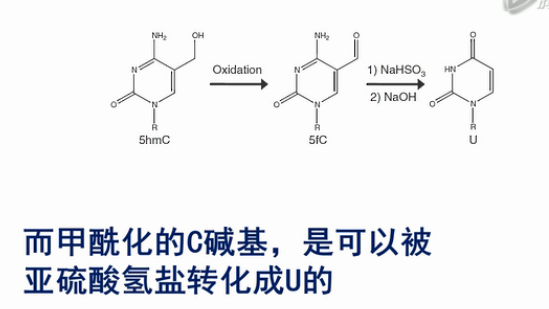
而甲基化的C,不会被转化成U。这样就把原来的羟甲基化的C和甲基化的C给区分开来了。
经研究表明,用高钌酸钾氧化的方法来氧化羟甲基化的C,其转化效率是94%左右。也就是说,每100个羟甲基化的C中,有94个会被高钌酸钾转化成甲酰化的C。并进一步被亚硫酸氢盐转化成U。
同时,原来的甲基货摊C,只有2.1%会被转化成甲酰化的C。
第二钟区分羟甲基化C的方法,是用糖基把羟甲基化的C给保护起来。然后用TET蛋白(Ten-eleven translocation methylcytosine dioxygenase 1),把甲基化的C转化成羟基化的C。
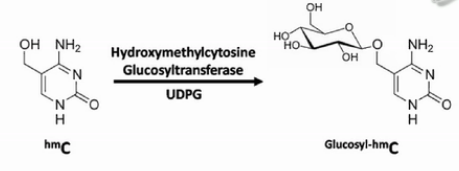
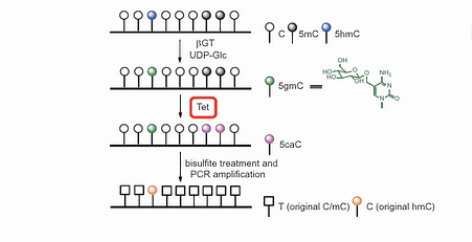
进一步将羟甲基化的C转化成甲酰化的C和羧基化的C。甲酰化的C和羧基化的C都可以被亚硫酸氢盐转化成U。
而之前被糖基化保护起来的羟甲基化的C,是不会被TET蛋白转化成甲酰化的C或者羧基化的C的。在亚硫酸氢盐的处理过程中,它还保持是C。并且在之后的PCR扩增产物中,也表现为C。
这样,就可以把羟甲基化的C,和甲基化的C,给区分开来。
用这个方法,没有甲基化的C,99.6%都被转化成了U。甲基化的C,97.7%都被转化成了U。而羟甲基化的C,只有8%被化成了U。
也就是说92%的羟甲基化的C得到了糖基的保护,还保持了C。
上述,就是目前2个区分羟甲基化的C和甲基化C的方法。
设置内参
在甲基化文库建程当中,亚硫酸氢盐对未甲基化的C的转化效率并不是100%,一般是在99%左右。
为了对实验的转化效率进行质量控制。一般会在转化实验当中加入内参对照品。
一般情况下,是用甲基化酶缺陷型的大肠杆菌,所生产出来的完全没有被甲基化的λ(噬菌体)DNA,或者pUC19(质粒)DNA做内参。来看一次实验当中C的转化效率。
一般情况下,实验当中是加入1%的完全没有甲基化的λ DNA做内参。
同样道理,也可以通过用甲基化酶处理过的,CpG岛完全被甲基化的DNA,来跟踪甲基化DNA对亚硫酸氢盐转化的抵抗效果。
数据分析
最后,我们来谈一下,甲基化测序后的数据处理。
因为亚硫酸氢盐处理过后,绝大部分的C都被转化成了T。这样,测出来的序列在和基因组进行对比的时侯,直接对比是对比不上的。
为了要进行比对,就要把基因组的碱基做两种转变。
第一种转变是把基因组上所有的C都改到T,再来和测序测到的序列来对比。这样,就可以把原来的链给对比上。
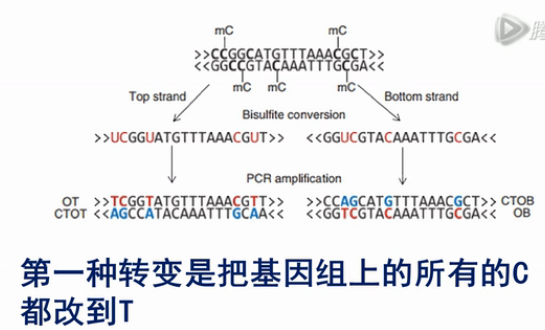
第二种转变,是把基因组上所有的G都变成A,这样才能和经过PCR得到的原样本链睥互补链对比得上。这样做的原因,是原样本链的互被链,它上面绝大部分的G,都被变成了A。所以,只有把(参考)基因组上的G,也都改成A,这样才能对比得上。
比对上之后,再来看哪些碱基是没有被转化的。这样,就可以确认这些碱基的甲基化修饰情况了。
再接下来,针对基因进行GO和Pathway的分析。在【陈巍学基因】第8期视频,RNA-seq当中,对GO和Pathway有详细的介绍,大家可以在优酷或者腾讯视频当中搜一下,看一下。
补充材料
DNA的甲基化分析,还有很多种方法,有兴趣的同学,可以在微信公众号【陈巍学基因】当中,回复“甲基”两个字,就可以看到7种其它分析DNA甲基化的方法。
7.Moleculo长测序
今天,会和大家谈一谈“Moleculo”测序方法。
Moleculo原来是美国的一家创业公司。这家公司开发了一种拼接长测序序列的方法。这个方法一经面世,就引起了Illumina的重视,Illumina马上出巨资,收购了这家公司。
在收购了Moleculo之后,Illumina把这个方法进行了优化。优化之后,以“TruSeq Synthetic
Long-Read DNA Library Kit”的形式,出现在Illumina的新产品当中。
在全新的基因组组装工作中,也就是我们通常所说的“De Novo”工作中,最核心的技术点,是能否得到大量的、长读长的序列。所以,得到长的读长序列,一直是做De novo工作的科学家所追求的有效技术手段。
另外,长读长的序列还可以帮助科学家来确定染色体单体的基因型。
Illumina标准的HiSeq/MiSeq测序方法,提供了一次给出大量序列的方法。它的序列,精度也很高,每个G的数据的测序成本也很低,但是,相对于De novo工作来说,它的读长还是不够长。
举例来说,Illumina旗下测序长度最长的MiSeq测序仪它的测序长度是:双端各300个碱基。那么,我们把这双端的300个碱基拼起来,中间交错100个碱基,可以得到一个500碱基的读长,
那么,我们要用500碱基读长的序列来组装一个和人类基因组大小相近的一个基因组,也就是单倍体长度为30亿个碱基长度的基因组,就相当于用筷子那么长(25厘米)的铁轨,来拼出一个京沪铁路(1300公里)。大家稍微想一想,就可以想出其中的难度。
Moleculo方法,它的巧妙点就是可以把Illumina不算太长的序列,拼接成一个一个10KB读长的序列,然后,再拼出基因组来。
接下来,我们就来介绍一下这个巧妙的办法。
第一步,分拆
首先是把长片段的基因组DNA,也就是40KB以上的长片段的基因组DNA,打断成10KB左右的DNA片段。
这个打断的过程,是用Covaris公司出品的g-TUBE方法来打断的。g-TUBE可以把长的基因组DNA,打断成5KB-20KB长度的片段。

打断了的DNA片段,末端大多数不是平齐的。接下来,就要用酶把这个末端给补平。
补平的过程,是用T4 DNA聚合酶、和Klenow聚合酶,两者的混合酶来进行补平。然后,再用T4 DNA寡核苷酸激酶,在5'端统一地加上磷酸基团。
补平之后,再用去掉了3'端外切酶活性的Klenow大片段聚合酶来进行处理。
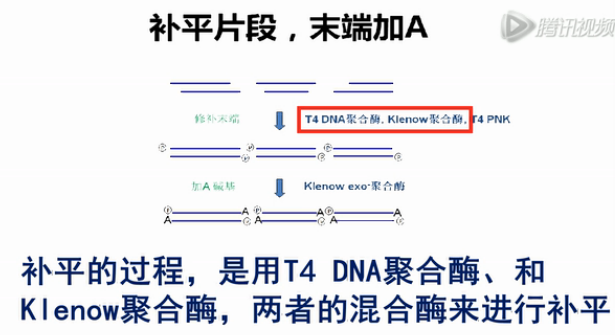
这样,可以在每个片段的两个3'端,都各加上一个A碱基。加好了A碱基之后再用连接酶,在DNA片段的两端连上第一步的PCR接头。
连好接头的DNA片段,走琼脂糖凝胶,切胶回收10KB左右的DNA片段。
回收下来的DNA片段,用qPCR进行精确定量。
第二步,扩增
用qPCR精确定量好之后的DNA片段,做成一个长PCR的Master Mix。
然后,把这个Master Mix分散到384孔PCR板里面,进行长PCR。

那么这里有一个注意点:就是如果是用来做De novo的文库,那么稀释到384孔的每一个小孔里,是3个fg(1 fg = 1 * 10^-15 g)的DNA。而如果是做染色体(单体)基因分型的,则是稀释到每个小孔75个fg的DNA。
之所以做De novo的这个PCR,要用更稀的模板,是因为,不希望一个小孔里面的片段,相互之间有交叠。
接下来,做长PCR在做长PCR的时侯,如果是用来做De novo的是做21个循环,而如果是做染色体基因分型的,则是做15个循环。
这个区别,是因为之前的两种反应,所加的起始模板量是不一样的。那么,现在要在PCR的环节当中,通过循环数的不一样,把DNA的最终产量,给拉平。
第三步,Nextera建库、测序
接下来,就用Nextera方法,对扩增好的片段,进行打断,并加上末端标签。
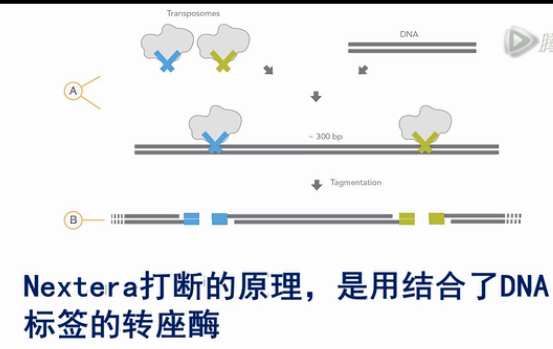
Nextera打断的原理,是用结合了DNA标签的转座酶,和之前扩增得到的10KB的DNA片段进行反应。
转座酶,一方面,会把长片段给切断成短的小片段。
另一方面,它也会把酶本身结合了的DNA标签,连在切出来的小片段DNA的末端上。
这个新加上的DNA标签,就成了接下来PCR扩增的引物结合序列。
再接下来,就是加入有P5、P7测序引物序列,同时带有Index序列的PCR引物,进行新的一轮PCR扩增。
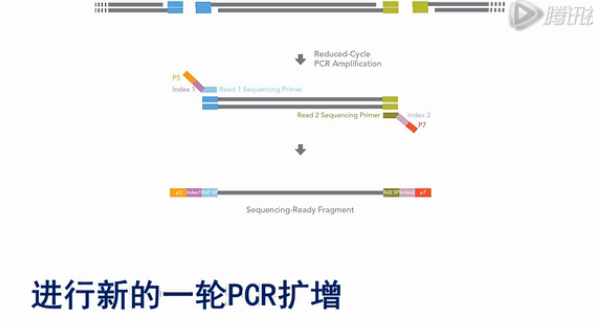
那么这一轮PCR扩增的结果,就会把Index序列,和P5、P7测序引物序列都加到扩增出来的DNA片段上。
这一轮的扩增完成之后,我们就得到的,就是384个带了完整的接头序列、Index序列的文库。
再接下来,就把这384个文库混合在一起,用柱子进行回收。
然后,就可以用Illumina测序仪进行测序了。
第四步,组装
测序完成之后,通过Index序列,把384个文库的序列可以分开,然后,分别进行组装。
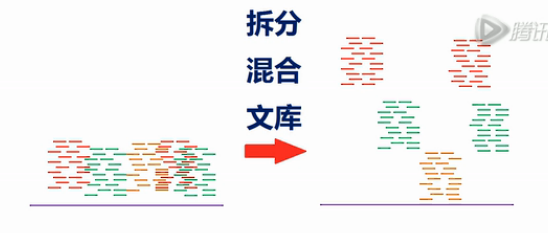
组装的结果,就是得到了许多个10KB的组装序列。
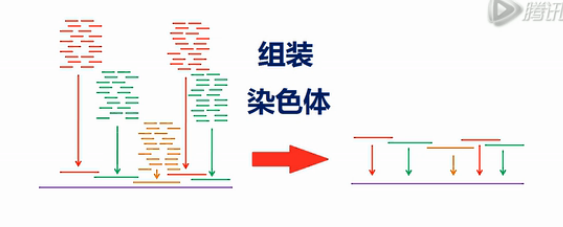
然后,可以用这10KB的组装序列,再去拼染色体的序列。
上面所说,就是Moleculo的合成长序列的测序方法。
要点总结
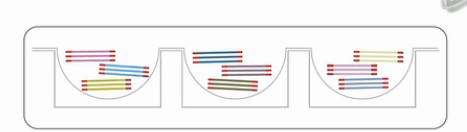
它的核心技术,就是把一个完整的基因组DNA,分成了384个小份。每一份中,又含了若干个10KB的DNA片段,而这一个小孔的中DNA片段,相互交叠的可能性很小。
所以,在重新组装的时侯,先组装成一个、一个10KB大小的片段。然后,再从10KB的片段,组装成染色体的序列。
这个,要比直接从几百个BP的序列,组装成染色体,要容易许多。
总的来说,Moleculo方法,就是把一个大难题,分解成2个相对容易解决的小问题,再进行分步地解决。最后,得到一个我们想要的结果。
以上是本期节目的全部内容,谢谢您的收看,我们下期节目再见。
8. Ribozero和方向性RNA文库
今天,会和大家谈一下RNA建库当中的RiboZero处理还有建方向性的RNA文库。
那么,我们先来说用RiboZero的方法,来处理总RNA之所以要用RiboZero方法来处理RNA。是因为在总RNA当中,大部分是核糖体RNA。而且这个比例高达95%左右而核糖体RNA在一个物种当中是高度保守的。所以,测核糖体RNA,一般情况下是没有什么研究意义的。
科学家测RNA,一般是想得到mRNA、还有Long non-coding RNA的变化信息。包括它的表达量变化,和结构上的变异信息。
所以,在RNA建库过程当中,很重要的一步就是要去除核糖体RNA。那么,要去除核糖体RNA最常用的方法,就是用带poly(T)探针的磁珠来和总RNA进行杂交。
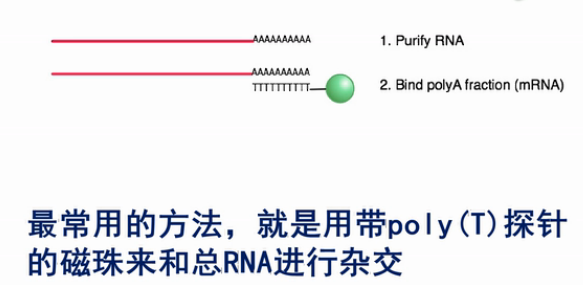
这样Poly(T)探针和mRNA上的Poly(A)尾巴结合。然后,用磁珠来回收这些吸附在探针上的、带poly(A)尾巴的mRNA,把mRNA洗脱下来之后,就可以进行下面的建库。
但是这个方法有一个缺点,就是它对总RNA质量的要求非常高。一般会要求总RNA的RIN值在8.0以上。如果总RNA有一定程度的降解,那么Poly(T)探针所能吸附到的,都是靠近mRNA的3’端的那些片段而mRNA的5’端的那些断片,就会大部分地被丢失。所以,测序得到的结果就会有很大的偏向性。

另外,如果是要测的是长链非编码RNA,也就是Longnon-coding RNA,也称作LncRNA,也是不能用Poly(T)方法来做的。因为大部分的LncRNA,它是没有Poly(A)尾巴的,所以它就不能用Poly(T)的探针来吸附。
RiboZero
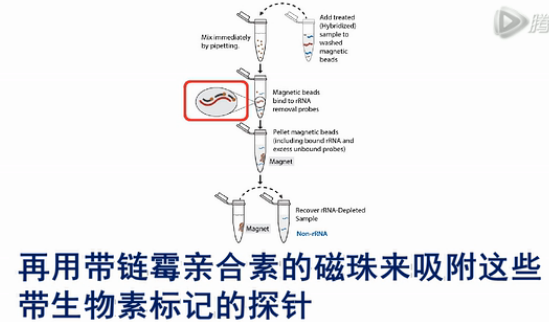
为了解决上述的问题,Illumina公司旗下的EpiCentre公司开发了RiboZero方法,来去除核糖体RNA。这个方法的原理不是通过探针来吸附带有Poly(A)尾巴的RNA序列。
而是倒过来,设计吸附核糖体RNA的探针,用探针来吸附核糖体RNA。再用带链霉亲合素的磁珠来吸附这些带生物素标记的探针。

最后磁珠被磁铁吸附在管壁上。
而其它的RNA,包括mRNA、LncRNA、和small RNA等RNA则留在上清液当中。
实验这样设计,就得到了2个结果。
第一点,就是对RNA样本的质量要求不再是很高。部分降解的RNA、或者降解程度很严重的RNA都可以用RiboZero的方法去除核糖体RNA。
最典型的是从石蜡样本归中回收的RNA样本,因为从石蜡样本中回收的RNA样本,它的质量是非常差的,之前是很难用来做测序的。现在有了RiboZero方法,就可以很方便地制备出文库来、并且进行测序。
第二点,就是那些不带Poly(A)尾巴的LncRNA,现在也可以被测序测到了。所以,现在市场上,大部分的LncRNA建库,都是通过RiboZero的方法,去除核糖体,接下来再进行建库。
但是RiboZero方法,它也有一个限制,就是每个物种的核糖体RNA的序列,它是有所不同的。
所以每种RiboZero的试剂盒,它其中的探针序列,都是有物种特异性的。EpiCentre公司开发了多个针对不同物种的RiboZero Kit。其中最常用的是针对:人、小鼠、大鼠的这个试剂盒。所以,科研客户在请测序公司进行RiboZero方法的建库、测序的时候,需要和测序公司确认所测的物种信息。
接下来,我们要介绍一下建定向的RNA库的方法。
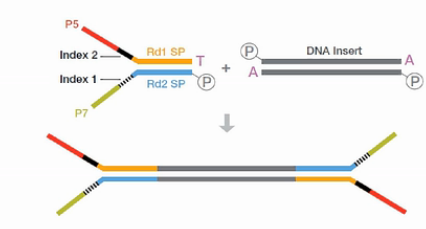
目前最常用的Truseq RNA建库方法,它是在双链cDNA的两端,对称地加上了两个Y型的接头,然后变成文库。

这个方法,它有一个缺点,就是它加接头的方向是对称的。所以测完序后,我们无法知道测出来的序列的方向性。也就是说,无法知道测到的是RNA的正义链,还是反义链。
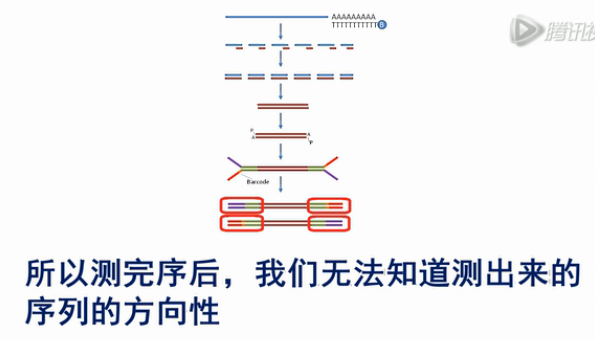
如果我们测的是人、小鼠之类的样本,那么问题不是很大。因为这些模式生物基因组序列,转录本序列,都是比较清楚的。
但是,如果我们是在测一些新的物种的时候,那么我们就不知道测到的是正义链,还是反义链了。
为了解决这个问题,科学家设计了多种方向性文库的建库方法。今天,我们就为大家介绍其中两种方向性的文库的建库方法。
掺U法
我们先来看这第一种方法。这种方法的原理,它是用掺入U碱基的办法,来标识cDNA的第二条链。
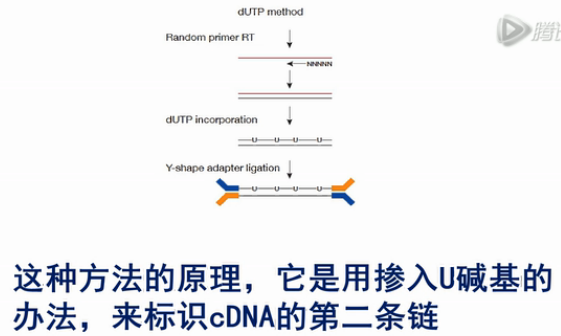
我们具体来看一下这个方法。
首先,它用常规的方法,从RNA上逆转录出第一链的cDNA。
然后,合成第二链时,所用的dNTP,它是特殊的。它用dUTP来代替了dTTP,用这样的dNTP来合成出的第二链,它当中就掺入了大量的U碱基。
而之前的第一链,是没有U碱基的。这样,第一链和第二链就有了差别。
接下来,在双链cDNA的两端接上Y型的接头。然后,用USER酶(Uracil-Specific Excision Reagent)进行消化。
那我们这里介绍一下USER酶。USER酶,它是一个混合酶。其中的尿嘧啶DNA糖基化酶(UDG)能够识别DNA链中的U碱基,并且把U碱基进行糖基化。接着,这个糖基化的U碱基从核酸链上切掉。这样核酸链上就型成了一个缺碱基的一个空位。接着,混合酶当中的核酸内切酶VIII就在脱碱基位点上把核酸链给切断掉。
刚才我们说了,在合成第二链的时候,是掺入了大量的U碱基那么这个双链的第二链就会被USER酶切得粉碎。也就是说cDNA的第二链被降解掉了。

降解发生之后,双链的文库就只剩下了一条链。而这条链的两头是接的不同序列的接头。
接下来进行PCR扩增。扩增出来的文库,保持了模板上的双个不对称的接头序列。
那么接下来,我们在测序的时候,测到就是有方向的文库了。
ScriptSeq法
接下来,我们介绍第二种建方向性文库的方法。
这个方法是Illumina公司的ScriptSeq方法。它的核心原理,是在加接头的时侯,左右两侧就加不同的接头。首先,它在合成第一链的cDNA的时候,它用的右侧引物,就是带了标签“A”

的接头。从这个接头延伸出来的cDNA链,很自然地在其右侧就连上了A接头序列。

接下来,把一个特殊的TTO引物(Termianl TaggingOligo )加进去。这个TTO引物的5’端是左侧的标签序列“B”。3’端是一连串随机序列,这些随机序列的作用是与刚才合成出来的第一链(cDNA)进行杂交。
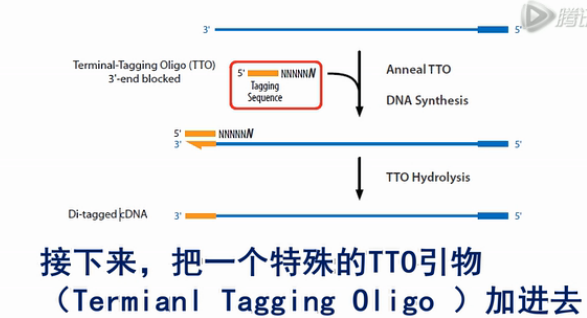
但是这个随机序列的3’端的最后一个碱基是一个双脱氧核苷酸,它的作用是不让这个TTO引物发生延伸反应。这个TTO引物与刚才合成的第一链cDNA杂交之后,第一链cDNA就在聚合酶的作用下,进一步延伸。延伸的结果就是把左侧的标签“B”也加到cDNA链上。
接下来,再用外侧的PCR引物对进行扩增,这对外侧的引物即带有一段与标签互补的序列。又带一段有与测序芯片上的接头互补的序列。这样扩增得到的产物,就是正式的文库了。
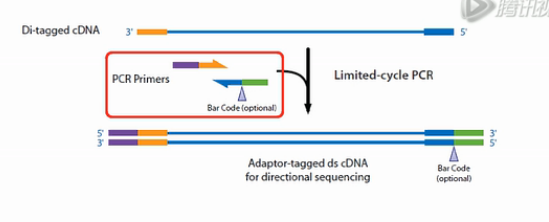
因为这个文库的左右是带了不同的标签的,所以这个文库,它测出来就是有方向性的。这样我们就得到了方向性的文库。
以上是本期视频的全部内容。
谢谢您的收看,我们下期节目再见。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号