

GlimmerHMM指南
GlimmerHMM指南
GlimmerHMM是一种De novo的新基因预测软件。
新基因发现基于Generalized Hidden Markov Model (GHMM)。
GlimmerHMM把一个基因看做几种特征序列(状态)的有序切换,这些特征序列包括内含子,基因间隔区,四种外显子(第一个外显子,中间的外显子,最后一个外显子,唯一的外显子),切换的过程形成马尔科夫链。
特征序列内部序列基于Nth-order interpolated Markov models (IMM) (N=8)处理。
描述基因中个特征序列转化关系的马尔科夫状态变化图如下:
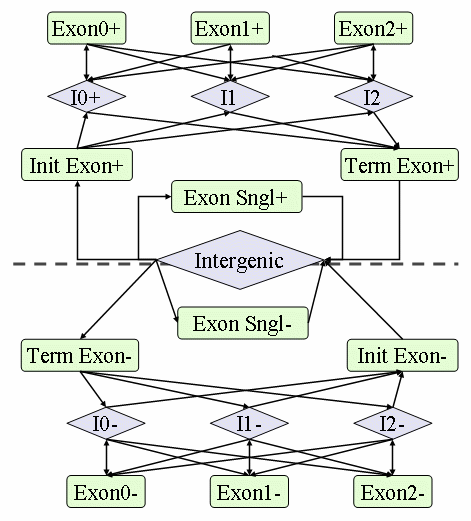
特征:
GlimmerHMM使用的模型基于以下几个假设,这些假设导致了GlimmerHMM的一些优点的不足:
- 假设每个基因都开始于起始密码子ATG
- 假设每个基因阅读框内除最后一个密码子外没有终止密码子(no in-frame stop codons)。
- 每个外显子与前一个外显子在同一个阅读框中。(翻译阅读时外显子间没有移框)
优点:GlimmerHMM的搜索范围下降,从而计算效率得以提高。
缺点:真正的移框外显子(genuine frame shifts)无法被检测到。
软件精度
参看官网数据
总体看比Genscan略好
安装
前往官网ftp下载最新版本,解压后即可使用。
Training
由于GlimmerHMM使用的数学模型是HMM,因此需要一个已经研究清楚的物种进行training(学习)之后再对新物种进行prediction(预测),用于trainning的物种应该和需要预测的物种具有较近的亲缘关系。
Input
GHMM的Training需要两个文件输入,即:
- mfasta_file
包含序列的fasta文件
>seq1
AGTCGTCGCTAGCTAGCTAGCATCGAGTCTTTTCGATCGAGGACTAGACTT
CTAGCTAGCTAGCATAGCATACGAGCATATCGGTCATGAGACTGATTGGGC
>seq2
TTTAGCTAGCTAGCATAGCATACGAGCATATCGGTAGACTGATTGGGTTTA
TGCGTTA
- exon_file
包含外显子位置信息,这个文件要求与mfasta_file一致:序列名称一致,位置序号正确指代mfasta_file的序列,不同的序列之间用空行隔开。
seq1 5 15
seq1 20 34
seq1 50 48
seq1 45 36
seq2 17 20
这个例子中,序列sep1具有两个基因,第一个在先导链上(the direct strand)后一个在互补链上(the complementary strand),每个基因有两个外显子。这里有一份真实的mfasta_file和exon_file,可用于理解exon_file,即后续程序的测试文件。
Training数据获取
一般来说,Trainning数据的获取主要有:
- 已经有良好实验背景的该物种基因信息(理想状况,一般不会太多)
- 从非冗余蛋白数据库(nr)中搜索能够map到基因组上的长ORFs(大于500bp),获取外显子位置信息 (比较常见)
Trainning
training的命令结构如下:
GlimmerHMM/train/trainGlimmerHMM <mfasta_file> <exon_file> [optional_parameters]
如果训练集中序列太少,程序会自动警告并且退出,默认情况下要求至少50个具有完整起始密码子(ATG)和终止密码子(TAA/TAG/TGA)的基因在训练集中。
[optional_parameters]中的参数在首次或常规运行中可以省略。
包括的可选项有:
-i----考虑isochores问题,例如:存在两种isochores,一种gc含量0-40%,另一种40-100%,则写为-i 0,40,100默认为-i 0,100
(isochore是指对温血动物的基因组片段做密度梯度离心实验时,发现的有的基因在离心管中聚集在一层,如同等高(体)线(isochore),这些基因的GC含量被认为相对均匀,长度大多大于300kb,这类DNA片段被命名为isochore。被认为是与脊椎动物的温血、冷血与否有关。)
-
-f -l -n -v分别是在已知上游UTR区、下游UTR区、基因间隔区、基因群周边序列的序列特征值的情况下输入特征值。(一般不需要填写) -
-b为splice位点简历1阶或2阶马尔科夫链(默认为1阶)
一般情况请直接输入
GlimmerHMM/train/trainGlimmerHMM <mfasta_file> <exon_file>
Trainning后的手动调整:
一般情况下,Trainning后用户不需要进行手动调整,但是手动调整可以一定程度上提高精度。这是因为,一些Trainning系统中的必要参数对于不同的物种有所区别,而系统并不知道(比如,基因间隔区的平均长度),因此,适当调整这些参数能够提升精度。
Majoros and Salzberg 2004描述了一种梯度渐进修改参数的方法慢慢提升精度。
除了提高精度外,修改参数还有很多意义。选择适当的假阳性和假阴性的容忍率,可以解决大量假阳性事件或者预测量太少的问题,从而在不减少太多预测结果的情况下提高准确率。例如,GlimmerHMM在预测一个剪切位点真实与否的时候会权衡假阳性概率,这会使得你错过一些位点,默认情况下这个假阳性的threshold是低于1%。你可以在调整GHMM的这些参数。
完成trainGlimmerHMM后,系统会在training产生的目录(就在工作目录下)下产生一个log文件来记录这次Trainning后GHMM自动产生的参数。
这个参数文件为train_0_100.cfg(前文Trainning时isochores没有被考虑时文件名是这样的,否则每个isochore会产生一个对应的cfg文件)每个可以改变的参数在这个文件中为一行,下表是参数和参数的描述(翻译后可能会失真,请阅读英文翻译)。
| line in the .cfg file | Description |
|---|---|
| acceptor_threshold value | acceptor site threshold value; the false negative/false positive rates for different acceptor thresholds can be consulted from the false.acc file. |
| donor_threshold value | donor site threshold value; the false negative/false positive rates for different donor thresholds can be consulted from the false.don file. |
| ATG_threshold value | start codon threshold value; the false negative/false positive rates for different start codon thresholds can be consulted from the false.atg file. |
| Stop_threshold value | stop codon threshold value; the false negative/false positive rates for different stop codon thresholds can be consulted from the false.stop file. |
| split_penalty value | a factor penalizing the gene finder's tendency to split genes. |
| intergenic_val value | the minimum intergenic distance espected between genes. |
| intergenic_penalty value | a penalty factor for genes situated closer than the intergenic_val value. |
| MeanIntergen value | the average size of intergenic regions. |
| BoostExon value | a factor to increase the sensitivity of exon prediction. |
| BoostSplice value | a factor to increase the score of good scoring splice sites. |
| BoostSgl value | a factor to increase the predicted number of single exon genes. |
| onlytga value | set this value to 1 if TGA is the only stop codon in the genome. |
| onlytaa value | set this value to 1 if TAA is the only stop codons in the genome. |
| onlytag value | set this value to 1 if TAG is the only stop codons in the genome. |
Running
GlimmerHMM的input文件包括两个,一个是fasta的序列文件,另一个是包含trainning结果的目录,一般来说,最好把他们放在一个工作目录下,这一步没有别的调整值(需要调整的都在*.cfg文件中完成)
GlimmerHMM/bin/glimmerhmm_linux fasta.file -d ./arabidopsis/

 浙公网安备 33010602011771号
浙公网安备 33010602011771号