Linux下Amber22安装与报错处理
如果没有购买amber,也可以安装ambertools进行分子分子动力学模拟。tools与amber的区别再于tools里没有高性能的pmemd,但tools里的sander也可以满足基本需求。有高性能(节约时间更快出结果)需求就需要购买正版!
-
安装环境配置
-
基础环境:按官网提示选择对应Linux发行版安装。传送门:https://ambermd.org/Installation.php
-
Ubuntu:apt -y update apt -y install tcsh make gcc gfortran flex bison patch bc wget flex bison patch bc wget
-
Centos7:yum -y install tcsh make gcc gcc-gfortran gcc-c++ which flex bison patch bc libXt-devel libXext-devel perl perl-ExtUtils-MakeMaker util-linux wget bzip2 bzip2-devel zlib-devel tar
-
-
必需:gcc、g++、gfortran 6.x版本以上,可以通过scl进行多版本gcc切换。
-
必需:cmake 3.x版本以上。如果自身系统的cmake是2.x版本,需要升级或编译3.x版本的cmake。传送门:https://cmake.org/download/
-
cmake编译
wget https://cmake.org/files/v3.20/cmake-3.20.0-rc2.tar.gz
tar -xvf cmake-3.20.0-rc2.tar.gz -C /usr/src/cmake3
cd /usr/src/cmake3/cmake-3.20.0-rc2
./configure --prefix=/usr/local/cmake3
make && make install
#添加软连接
ln -s /usr/local/cmake/bin/cmake /usr/bin/cmake
#或添加环境变量
export PATH=$PATH:/usr/local/cmake3/bin
-
-
cuda 环境 ,如果不需要使用GPU,可以忽略此。
注意cuda版本要与gcc的版本匹配!
![]()
-
并行环境:openmpi或在intel等提供的mpi。建议直接按官方提示配置并行环境。
-
-
安装步骤:
-
解压文件:
tar xvfj AmberTools22.tar.bz2 # (Extracts into an “amber22_src” directory.)
tar xvfj Amber22.tar.bz2 # (Only if you have licensed Amber 22!)
-
配置文件:
-
进入amber_scr/build文件夹,如果没有特殊需求直接按照默认,则直接执行:sudo ./run_cmake
-
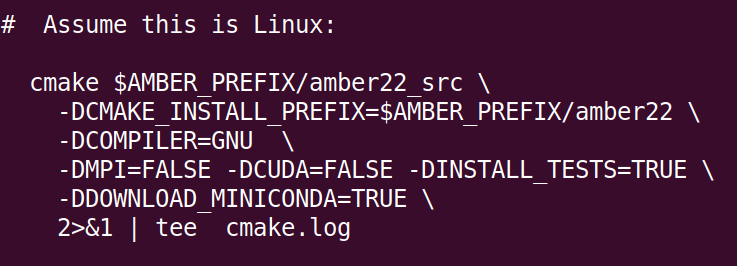
如果需要特殊配置,则编辑run_cmake文件,举例说明:
![]()
-
cuda需求:-DCUDA=FALSE改为TRUE
-
mpi需求:-DMPI=FALSE改为TRUE
-
如果需要开启其他模块,在该行末尾添加即可:比如添加qmmm计算模块quick:-DBUILD_QUICK=TRUE
-
-
-
执行编译:
-
sudo ./run_make 需要网络,完成后会有提示,可以把source那句拷贝下来,后面用得着。
-
sudo make install
-
添加环境变量进.bashrc:source /xxx/xxx/xxx/amber.sh(第一步完的拷贝的就在这里用!)
-
-
-
报错处理:
-
网络原因,miniconda.sh下载太慢,可以提前下载好拷贝进对应文件夹。
-
报错:
make[2]: ***[src/pmemd/src/xray/cuda/CMakeFiles/pmemd_xray_cuda.dir/src/xray/pmemd_xray_cuda_generated_BulkMaskGPU.cu.o]
Error 1 make[2]: Leaving directory
/home/nitin.bt.iith/Amber22/amber22_src/build' make[1]: *** [src/pmemd/src/xray/cuda/CMakeFiles/pmemd_xray_cuda.dir/all] Error 2 make[1]: Leaving directory/home/nitin.bt.iith/Amber22/amber22_src/build' make: *** [all] Error 2解决办法:
cd amber22_src/src/pmemd/src mv CMakeLists.txt CMakeLists.txt.original cp CMakeLists.txt.noxray CMakeLists.txt # rebuild cd amber22_src/build ./clean_build ./run_cmake make install
-
注意:gcc、g++、gfortran 版本尽量保持一致,不然后面的编译容易出错!
-


 浙公网安备 33010602011771号
浙公网安备 33010602011771号