


Seurat分析空间数据集(10x Visium 非HD)
内容参考官方教程
数据读取、数据预处理、基因表达可视化、降维,聚类和可视化、交互式绘图、识别空间可变特征、细分解剖区域、与单细胞数据集成、在Seurat中处理多个切片
关于 $10X$ 数据
\(10X\) 数据是指通过 \(10X Genomics\) 平台 产生的单细胞或单细胞核测序原始数据。
\(10X\) 的单细胞/单核测序数据和普通 \(RNA-seq\) 数据的最大区别是 每条测序 \(reads\) 前面都有“身份标签”:
Cell Barcode(CB) 标识来自哪一个单细胞/单核(相当于细胞身份证)
UMI(Unique Molecular Identifier) 标识转录本的独立分子,去除 PCR 扩增偏差用
Read 实际的转录本序列
\(10X\) 数据测序流程
一、10x 单细胞测序实验流程
1. 样本准备
得到组织 → 制备成单细胞悬液(或单核悬液,如果做 snRNA-seq)
控制细胞浓度,避免多个细胞进入同一微滴
2. 微滴包裹(Gel Bead-In-Emulsion,GEM)
细胞悬液 + 酶体系 + 条形码微珠(Gel Bead) 被分配进油相微滴中
理想情况:一个微滴中只有一个细胞 + 一颗条形码微珠
条形码微珠的核心结构(每颗微珠表面有大量寡核苷酸引物):
Cell Barcode(CB):标识细胞来源
UMI:标识转录本分子
Poly(dT):捕获 mRNA 的 poly(A) 尾巴
3. 细胞裂解 & mRNA 捕获
在微滴中,细胞被裂解 → mRNA 释放
mRNA 的 poly(A) 尾巴 与微珠上的 poly(dT) 结合
此时每个 mRNA 分子会被打上 Cell Barcode + UMI
4. 逆转录(RT)
在微滴中进行逆转录反应
mRNA → cDNA
每条 cDNA 上已经带有:Cell Barcode + UMI
5. 打破微滴 & 文库构建
破乳(把油相微滴打破)
收集所有 cDNA
PCR 扩增 → 加测序接头 → 得到测序文库
6. 高通量测序(Illumina 平台)
通常采用 Illumina 双端测序(比如 PE150):
Read 1:包含 UMI + Cell Barcode 信息
Read 2:包含 cDNA 序列(即转录本片段)
Index Read:文库 index
二、10x 单细胞数据处理流程
测序后得到 FASTQ 文件,接下来用 Cell Ranger 软件进行处理:
1. demultiplex
从测序数据中识别出每个细胞的 Cell Barcode
丢弃低质量的条形码
2. 比对(alignment)
将 cDNA 序列(Read2)比对到参考基因组/转录组
snRNA-seq 时会加 --include-introns
3. UMI 去重
利用 UMI 去除 PCR 扩增产生的冗余计数
得到真实的转录本拷贝数
4. 生成表达矩阵
输出一个稀疏矩阵:
行:基因
列:细胞
值:UMI counts(基因在该细胞中的表达量)
在 \(Seurat\) 里看到的列名(细胞名),来源于 \(10X\) 数据的 \(Cell Barcode(CB)\) ,而不是 \(UMI\)。如AAACCTGAGCGAAAC-1,-1 是技术性标记(表示这是第一个文库 \(lane\),如果多个文库合并会出现 -2, -3 …)\(UMI\) 并不出现在矩阵的细胞名里,它只在计算 \(UMI count\) 时起作用
\(UMI\) 去重原理
PCR 扩增会造成 同一个 mRNA 被复制成很多条 reads,如果不校正,就会错误地认为表达量更高。
UMI(Unique Molecular Identifier)就是用来解决这个问题的。
例子:
假设某个细胞里有一个基因 GeneA 的 mRNA 分子,真实数量是 2 个分子。
实验流程:
每个 mRNA 分子在捕获时都会被打上一个随机的 UMI 标签(例如长度 10bp,可以产生约 1 百万种不同标签)。
第一个分子 → UMI = ATCGTACGTA
第二个分子 → UMI = GCTAGTTCAG
PCR 扩增
第一个分子扩增出 100 条 reads(都带着同样的 UMI ATCGTACGTA)
第二个分子扩增出 80 条 reads(UMI GCTAGTTCAG)
测序比对 + 去重
Cell Ranger 或其他分析工具会统计:
UMI = ATCGTACGTA → 1 个分子
UMI = GCTAGTTCAG → 1 个分子
最后得出 GeneA 在该细胞里的 真实 UMI count = 2
\(10X\) 结果数据组成
1. barcodes.tsv
存储的是所有 细胞条形码(Cell Barcodes)
每一行对应一个条形码(即一个细胞或一个微滴)
示例:AAACCTGAGCGAAAC-1
-1 表示这是第 1 个 library/lane,如果你合并多个样本,可能会出现 -2, -3 ...
在矩阵里,对应列名(细胞名)
2. genes.tsv(或新版叫 features.tsv)
存储的是 基因信息
两列或三列:
第一列:基因 ID(如 ENSG00000121410)
第二列:基因符号(如 TSPAN6)
第三列(如果有):基因类别(如 Gene Expression,snRNA-seq 时可能还会有 Antibody Capture、CRISPR Guide Capture 等)
示例:ENSG00000243485 MIR1302-2HG Gene Expression
在矩阵里,对应行名(基因名/ID)
3. matrix.mtx
存储的是 表达矩阵(稀疏矩阵,Matrix Market 格式)
行 → 基因 列 → 细胞 值 → 该细胞中该基因的 UMI 计数
之所以用稀疏矩阵,是因为大部分基因在单细胞中是零表达
示例(伪数据,真实文件是稀疏格式):
%MatrixMarket matrix coordinate integer general
20000 5000 1234567 ← 20000个基因, 5000个细胞, 1234567个非零值
1 1 2 ← 基因1在细胞1里有2个UMI
5 1 1 ← 基因5在细胞1里有1个UMI
2 2 3 ← 基因2在细胞2里有3个UMI
...
\(10X\) 空间转录组(\(Visium\) 平台):
1. 普通 10x 单细胞/单核测序(scRNA-seq/snRNA-seq):
样本被打散成单细胞悬液
丢失了空间位置信息(只知道某个细胞表达了哪些基因,不知道它原来在组织里的位置)
2. 10x 空间转录组(Visium 平台)
不需要消化成单细胞,而是直接把组织切片放在玻片上
玻片表面有很多固定的“捕获点”(spots),每个 spot 上带有空间坐标的 barcode
每个 spot 捕获范围大约 55 μm,通常包含 多个细胞的 RNA
所以最后得到的不是“单细胞表达矩阵”,而是“空间 spot 表达矩阵”+ “空间坐标信息”
\(Visium\) 实验流程
(1) 样本制备
将组织切片(石蜡切片/冷冻切片)放在 Visium Slide(玻片) 上
每张玻片有 4 个 capture area,每个区域约 6.5×6.5 mm²
(2) Spot 捕获探针阵列
每个 capture area 上有 ~5000–6000 个 spots(捕获点)
每个 spot 直径 ~55 μm,间距 ~100 μm(Visium HD 分辨率更高)
每个 spot 上的寡核苷酸探针包含:
Spatial Barcode(空间条形码) → 记录 spot 的位置
UMI(Unique Molecular Identifier) → 区分不同分子
Poly(dT) → 捕获 poly(A) 尾的 mRNA
(3) 组织处理与 RNA 捕获
在玻片上固定组织 → 透化(permeabilization)
组织中 RNA 分子释放并被 spot 上的探针捕获
这样,每条 RNA 就带有它来自哪个 spot 的空间条形码
(4) 逆转录 & 文库构建
在玻片上直接进行逆转录(RT) → mRNA → cDNA
加入 UMI & Spatial Barcode 信息
然后文库构建 + 高通量测序(Illumina)
(5) HE 染色 & 成像
同一张切片会做 H&E 染色,拍摄组织形态学图像
后续会将 基因表达矩阵 与 HE 图像进行对齐(alignment)
Visium得到的结果中,分析需要用到的数据以及各个数据存储的内容在代码注释中
Visium 里的 spot ≠ 单细胞测序里的微滴,它们虽然都依赖“条形码”来标记 RNA 来源,但原理完全不同。
1. 普通 10x 单细胞 (scRNA-seq)
载体:微滴(droplet in oil)
条形码来源:每个微滴里包裹一颗 Gel Bead(上面带 Cell Barcode + UMI)
原理:
一个细胞 + 一颗微珠 → 在油滴里裂解
释放的 mRNA 被捕获并加上“Cell Barcode”
特点:
一个微滴 ≈ 一个细胞(理论上)
所以表达矩阵列名(Cell Barcode) = 单细胞
2. 10x Visium 空间转录组
载体:固定玻片上的 spot 捕获探针阵列(不是油滴)
条形码来源:每个 spot 上的寡核苷酸探针带有 空间条形码 + UMI
原理:
把组织切片直接放在玻片上
组织透化 → 释放的 RNA 向下扩散,被 spot 捕获
所以每个 spot 捕获的是 该区域内多个细胞的混合 RNA
特点:
一个 spot ≠ 一个细胞
一个 spot ~55 μm,往往覆盖 1–10+ 细胞
表达矩阵列名(spot Barcode) = 空间位置,而不是单个细胞
整体代码
#官方教程
#https://satijalab.org/seurat/articles/spatial_vignette
library("Seurat")
#devtools::install_github('satijalab/seurat-data')
library("SeuratData")
library("ggplot2")
library("patchwork")
library("dplyr")
setwd('E:\\AAA大创\\代码注释版\\暑假练习\\空间转录组')
# 读取 Visium 数据
#这里使用的是示例数据,InstallData可以下载加安装,如果下载不下来的话
#可以在https://seurat.nygenome.org/src/contrib/stxBrain.SeuratData_0.1.2.tar.gz下载后使用install.packages安装
#install.packages("E:\\AAA大创\\代码注释版\\暑假练习\\空间转录组\\stxBrain.SeuratData_0.1.2.tar.gz", repos = NULL, type = "source")
InstallData("stxBrain")
brain <- LoadData("stxBrain", type = "anterior1")
#anterior1是stxBrain中小鼠前脑第一张切片的数据,类似的还有anterior2,posterior1
#如果不使用示例数据,而是去10x官网下载的话,需要下载Feature/barcode matrix HDF5 (filtered)
#和Spatial imaging data至少两个文件,将GZ文件解压到和h5同一个目录后运行以下代码后即可读取
#注意新的HD文件有些变化,此处的方法无法使用
brain <- Load10X_Spatial(
data.dir = "E:\\AAA大创\\代码注释版\\暑假练习\\空间转录组\\data",
filename = "CytAssist_FFPE_Human_Colon_Post_Xenium_Rep1_filtered_feature_bc_matrix.h5"
)
#关于各种文件内容:
#h5保存了单细胞/空间条形码的基因表达矩阵
#tissue_hires_image.png为高分辨率组织切片图像。
#tissue_lowres_image.png为低分辨率图像,加载和渲染更快,默认时常用
#aligned_tissue_image.jpg为对齐后的组织图像,一般是把组织切片图像和探针阵列对齐后的结果。
#aligned_fiducials.jpg为对齐时使用的参照点
#Fiducials 是 Visium 芯片边缘上的小白点,用来帮助软件自动对齐组织切片和探针阵列。
#detected_tissue_image.jpg为自动识别组织区域后的结果图像,会标出哪些区域被判定为“组织覆盖”,哪些是背景。
#cytassist_image.tiff为扫描仪捕获的组织图像。是后续对齐、生成 aligned_tissue_image 的原始图像来源。
# scalefactors_json.json存放不同图像分辨率与 spot 坐标系统之间的比例系数。
#用于把 barcode 的坐标映射到 hires 或 lowres 图像上。
#tissue_positions.csv(或 .csv.gz)每个 spot 条形码的空间坐标。
#包含条形码 ID 是否落在组织区域 像素坐标(x,y)
#spatial_enrichment.csv 组织区域富集分析相关的信息
plot1 <- VlnPlot(brain, features = "nCount_Spatial", pt.size = 0.1) + NoLegend()
#pt.size = 0.1:每个点的大小(单个 spot 的数值显示为散点,叠加在小提琴图上)。
plot2 <- SpatialFeaturePlot(brain, features = "nCount_Spatial") + theme(legend.position = "right")
pdf(file="1.nCount_Spatial.pdf",width=20,height=12)
wrap_plots(plot1, plot2)
#前面生成的小提琴图和空间分布图拼到同一画布。
dev.off()
brain <- SCTransform(brain, assay = "Spatial", verbose = FALSE)
#assay = "Spatial":指定输入数据来源于 "Spatial" assay(Visium 数据导入后默认的 assay 名)。
#verbose = FALSE:关闭运行过程的打印信息(让输出更干净)。
#因为空间转录组测序时不同位置的细胞密度不同,因此不同位置表达量的差异不单单表示表达量的高低,
#此时使用LogNormalize()等标准化方法会导致密度等因素的丧失,这里采用SCTransform的方式进行归一化
pdf(file="2.SCTransform.pdf",width=20,height=12)
SpatialFeaturePlot(brain, features = c("Hpca", "Ttr"))
#默认使用brain中 "SCT" assay 的数据,除非你用 assay= 参数指定
#features = c("Hpca", "Ttr"):要展示的基因。
dev.off()
#另一种输出方式
#plot <- SpatialFeaturePlot(brain, features = c("Ttr")) + theme(legend.text = element_text(size = 0), legend.title = element_text(size = 20), legend.key.size = unit(1, "cm"))
#theme修改图例样式:
#legend.text = element_text(size = 0)图例数值文字大小设为 0。
#legend.title = element_text(size = 20)图例标题(Ttr)字号设置为 20。
#legend.key.size = unit(1, "cm")图例色条的大小设为 1 cm 高。
#jpeg(filename = "../output/images/spatial_vignette_ttr.jpg", height = 700, width = 1200, quality = 50)
#quality = 50为JPG 压缩质,范围 0–100,数值越大越清晰但文件越大
#print(plot)
#dev.off()
#SpatialFeaturePlot参数设置
p1 <- SpatialFeaturePlot(brain, features = "Ttr", pt.size.factor = 1)
#pt.size.factor = 1 控制spot大小
p2 <- SpatialFeaturePlot(brain, features = "Ttr", alpha = c(0.1, 1))
#控制 spot 的 透明度。表达值低的 spot透明度 = 0.1,表达值高的 spot透明度 = 1(完全不透明)
pdf(file="3.SCTransform.pdf",width=20,height=12)
p1 + p2
dev.off()
#使用UMAP可视化聚类结果
brain <- RunPCA(brain, assay = "SCT", verbose = FALSE)
brain <- FindNeighbors(brain, reduction = "pca", dims = 1:30)
brain <- FindClusters(brain, verbose = FALSE)
brain <- RunUMAP(brain, reduction = "pca", dims = 1:30)
p1 <- DimPlot(brain, reduction = "umap", label = TRUE)
p2 <- SpatialDimPlot(brain, label = TRUE, label.size = 3)
#使用label = TRUE来在切片图中进行标注
pdf(file="4.UMAP.pdf",width=20,height=12)
p1 + p2
#输出时可能会有好几种报错方式,大概率是Seurat和ggplot2以及patchwork这几个包的问题
#可以更新一下尝试解决
dev.off()
pdf(file="5.CellsByIdentities.pdf",width=20,height=12)
SpatialDimPlot(brain, cells.highlight = CellsByIdentities(object = brain, idents = c(2, 1, 4, 3, 5, 8)), facet.highlight = TRUE, ncol = 3)
#要提取 簇(cluster)编号为 2, 1, 4, 3, 5, 8 的细胞。在空间图上被 高亮显示。
#facet.highlight控制是否单独绘制每个高亮细胞簇的分面图
#SpatialDimPlot(brain, cells.highlight = CellsByIdentities(object = brain, idents = c(2, 1, 4, 3, 5, 8)), facet.highlight = FALSE, ncol = 3, cols.highlight = c("red", "blue", "green", "orange", "purple", "cyan", 'yellow'))
#放在同一张图的写法,注意提供颜色参数
dev.off()
#交互式窗口
SpatialDimPlot(brain, interactive = TRUE)
#可查看对应spot所属Group
SpatialFeaturePlot(brain, features = "Ttr", interactive = TRUE)
#可交互式调整透明度,点的大小和颜色方案
LinkedDimPlot(brain)
#同时显示UMAP图和组织图像,可以通过点击umap图中区域来高亮组织图像中对应区域,也可通过点击组织区域来高亮对应区域。
#识别空间可变特征
#基于先前的聚类结果进行识别
de_markers <- FindMarkers(brain, ident.1 = 5, ident.2 = 4)
#检测 cluster 5 相对于 cluster 4 显著差异的基因。
SpatialFeaturePlot(object = brain, features = rownames(de_markers)[1:3], alpha = c(0.1, 1), ncol = 3)
#绘制差异分析结果里排名前3的基因在切片上的空间表达分布
#注意默认排序方式是按照p值从小到大排序,如有需要请提前对de_markers进行修改
#alpha = c(0.1, 1)控制 spot 的透明度映射,ncol = 3绘图时一行展示 3 张图
#寻找那些在组织空间分布上不是随机的,而是存在空间模式的基因
brain <- FindSpatiallyVariableFeatures(brain, assay = "SCT", features = VariableFeatures(brain)[1:1000], selection.method = "moransi")
#从对象里取前 1000 个高变基因,使用 Moran’s I 指数 来度量空间自相关。method默认为markvariogram
#跑的慢的话可以试试install.packages('Rfast2')
#VariableFeatures来自SCTransform()的分析结果
#各种报错的可能是各种包版本问题,可以试着把R和R包都更新一波
#结果里面(brain[["SCT"]]@meta.features)只有指定的基因(前1000个)有结果,其余为NA
top.features <- head(SpatiallyVariableFeatures(brain, selection.method = "moransi"), 6)
#对象里提取按 Moran’s I 排序的前六个空间可变基因
pdf(file="6.SpatialFeaturePlot.pdf",width=20,height=12)
SpatialFeaturePlot(brain, features = top.features, ncol = 3, alpha = c(0.1, 1))
#展示得到的基因
dev.off()
##细分解剖区域
cortex <- subset(brain, idents = c(1, 2, 3, 4, 6, 7))
#提取特定Cluster的spot
spatial_coords <- GetTissueCoordinates(cortex)
#用于从空间对象中提取坐标,屏蔽了底层 VisiumV1 和 VisiumV2 类的差异
cortex$imagerow <- spatial_coords[colnames(cortex), "x"] # 注意细胞ID匹配和坐标方向
cortex$imagecol <- spatial_coords[colnames(cortex), "y"]
#将空间坐标添加到 Seurat 对象的元数据中
SpatialDimPlot(cortex,cells.highlight = WhichCells(cortex, expression = imagerow > 5000 & imagecol > 7500))
#可视化筛选区域,辅助边界确定
#以下代码为根据目标区域对解剖区域进行划分
cortex <- subset(cortex, imagerow > 6000 & imagecol > 6000, invert = TRUE)
cortex <- subset(cortex, imagerow > 5000 & imagecol > 7500, invert = TRUE)
#出现大量警告信息只是提示没做合法性验证,不会影响结果
#注意这里追加了invert=TRUE,那么这里的代码不是筛选对应的细胞,而是去掉对应位置的细胞
pdf(file="7.Subset.pdf",width=20,height=12)
p1 <- SpatialDimPlot(cortex, crop = TRUE, label = TRUE)
p2 <- SpatialDimPlot(cortex, crop = FALSE, label = TRUE, pt.size.factor = 1, label.size = 3)
#crop = TRUE:只显示组织区域(切掉空白边缘)。
#crop = FALSE:显示整个载玻片区域(包括背景)。
p1 + p2
dev.off()
##整合单细胞数据
#贴一个从官网找到的数据链接https://www.dropbox.com/s/cuowvm4vrf65pvq/allen_cortex.rds?dl=1
allen_reference <- readRDS("./data/allen_cortex.rds")
#从 RDS 文件中读取 Allen 研究所提供的皮层单细胞 scRNA-seq 数据
#使用的数据不一定是同一块组织的,因为后续会进行匹配,但是推荐同一物种+同一组织+相同测序平台
library(dplyr)
#BiocManager::install('glmGamPoi')
#系统提示安装后能加快一些速度
allen_reference <- SCTransform(allen_reference, ncells = 3000, verbose = FALSE) %>%
RunPCA(verbose = FALSE) %>%
RunUMAP(dims = 1:30)
#SCTransform:使用 sctransform 归一化方法对参考 scRNA-seq 数据进行标准化。
#ncells=3000:对所有细胞做归一化,但只用 3000 个细胞来构建噪声模型。噪声模型用于识别哪些信息是噪声
#save.image("E:/AAA大创/代码注释版/暑假练习/空间转录组/umap.RData")
#跑完及时存储,避免反复跑
cortex <- SCTransform(cortex, assay = "Spatial", verbose = FALSE) %>%
RunPCA(verbose = FALSE)
#对cortex进行分析
# the annotation is stored in the 'subclass' column of object metadata
pdf(file="8.allen_reference.pdf",width=20,height=12)
DimPlot(allen_reference, group.by = "subclass", label = TRUE)
#按照allen_reference数据中的subclass资料进行分组展示
dev.off()
anchors <- FindTransferAnchors(reference = allen_reference, query = cortex, normalization.method = "SCT")
#作用:在 allen_reference(参考 scRNA-seq 数据)和 cortex(空间转录组数据)之间寻找“锚点(anchors)”
#锚点:就是两组数据中表达模式相似的细胞对,用来对齐和传递信息。
#normalization.method = "SCT":表示使用 SCTransform 标准化方式来进行匹配。
prediction.assay = TRUE
#默认情况下,TransferData() 会把参考数据的标签直接写入 query 对象的 meta.data
#但是如果你设置 prediction.assay = TRUE,它会在 query 对象中新建一个名为 "predictions"的 Assay,存储概率矩阵:
#表示该细胞属于对应标签的概率(介于 0~1)。
predictions.assay <- TransferData(anchorset = anchors, refdata = allen_reference$subclass, prediction.assay = TRUE,
weight.reduction = cortex[["pca"]], dims = 1:30)
#作用:把参考数据中的注释(subclass,也就是细胞亚类)转移到 cortex 上。
#anchors: 上一步找好的锚点。
#refdata = allen_reference$subclass: 把参考数据中 subclass 注释当作要转移的标签。
#prediction.assay = TRUE: 结果存放在一个新的 assay 里(名为 "predictions")。
#weight.reduction = cortex[["pca"]]: 在 PCA 降维空间中做标签转移。
#dims = 1:30: 使用前 30 个主成分。
cortex[["predictions"]] <- predictions.assay
DefaultAssay(cortex) <- "predictions"
#把预测结果存到 cortex 里,并且设置 "predictions" 作为默认 assay(后续可直接调用预测分数)。
pdf(file="9.cortex_predictioins.pdf",width=20,height=12)
SpatialFeaturePlot(cortex, features = c("L2/3 IT", "L4"), pt.size.factor = 1.6, ncol = 2, crop = TRUE)
#展示L2/3 IT、L4两组,同时使用crop裁剪掉多余的空白区域。
dev.off()
cortex <- FindSpatiallyVariableFeatures(cortex, assay = "predictions", selection.method = "moransi",
features = rownames(cortex), r.metric = 5, slot = "data")
#在 cortex Seurat 对象里,寻找空间上变化显著的基因/特征。这里指定使用Moran’s I来选择空间可变特征。
#使用的是存放在 predictions assay 里的数据
#features指定要测试的特征,即"Vip""Lamp5""Sst""Sncg"等
#r.metric规定局部邻域多少范围内的点纳入统计
#slot = "data"使用 assay 中的 data 层而不是count或者scale.data层,虽然是概率矩阵,但是这两层仍然存在
top.clusters <- head(SpatiallyVariableFeatures(cortex, selection.method = "moransi"), 4)
#空间可变特征里取前4个最显著的特征
pdf(file="10.predictioins_SpatialPlot.pdf",width=20,height=12)
SpatialFeaturePlot(object = cortex, features = top.clusters, ncol = 2)
dev.off()
#展示选出的前4个可变特征的空间分布
pdf(file="11.predictioins_SpatialFeaturePlot.pdf",width=20,height=12)
SpatialFeaturePlot(cortex, features = c("Astro", "L2/3 IT", "L4", "L5 PT", "L5 IT", "L6 CT", "L6 IT",
"L6b", "Oligo"), pt.size.factor = 1, ncol = 2, crop = FALSE, alpha = c(0.1, 1))
#展示指定类型的空间分布
dev.off()
##Seurat处理多个切片
brain2 <- LoadData("stxBrain", type = "posterior1")
#数据集包含另一个与大脑另一半相对应的切片。
brain2 <- SCTransform(brain2, assay = "Spatial", verbose = FALSE)
#合并两个切片数据
brain.merge <- merge(brain, brain2)
DefaultAssay(brain.merge) <- "SCT"
VariableFeatures(brain.merge) <- c(VariableFeatures(brain), VariableFeatures(brain2))
brain.merge <- RunPCA(brain.merge, verbose = FALSE)
brain.merge <- FindNeighbors(brain.merge, dims = 1:30)
brain.merge <- FindClusters(brain.merge, verbose = FALSE)
brain.merge <- RunUMAP(brain.merge, dims = 1:30)
DimPlot(brain.merge, reduction = "umap", group.by = c("ident", "orig.ident"))
#ident:按照聚类结果上色
#orig.ident:按照样本来源上色
pdf(file="12.merge_SpatialDimPlot.pdf",width=20,height=12)
SpatialDimPlot(brain.merge)
dev.off()
pdf(file="13.merge_SpatialFeaturePlot.pdf",width=20,height=12)
SpatialFeaturePlot(brain.merge, features = c("Hpca", "Plp1"))
#SpatialDimPlot()和 SpatialFeaturePlot()的默认排版是将不同切片绘制为列,将不同分组/要素绘制为行。
dev.off()
一、数据读取
Show Code
# 读取 Visium 数据
#这里使用的是示例数据,InstallData可以下载加安装,如果下载不下来的话
#可以在https://seurat.nygenome.org/src/contrib/stxBrain.SeuratData_0.1.2.tar.gz下载后使用install.packages安装
#install.packages("E:\\AAA大创\\代码注释版\\暑假练习\\空间转录组\\stxBrain.SeuratData_0.1.2.tar.gz", repos = NULL, type = "source")
InstallData("stxBrain")
brain <- LoadData("stxBrain", type = "anterior1")
#anterior1是stxBrain中小鼠前脑第一张切片的数据,类似的还有anterior2,posterior1
#如果不使用示例数据,而是去10x官网下载的话,需要下载Feature/barcode matrix HDF5 (filtered)
#和Spatial imaging data至少两个文件,将GZ文件解压到和h5同一个目录后运行以下代码后即可读取
#注意新的HD文件有些变化,此处的方法无法使用
brain <- Load10X_Spatial(
data.dir = "E:\\AAA大创\\代码注释版\\暑假练习\\空间转录组\\data",
filename = "CytAssist_FFPE_Human_Colon_Post_Xenium_Rep1_filtered_feature_bc_matrix.h5"
)
#关于各种文件内容:
#h5保存了单细胞/空间条形码的基因表达矩阵
#tissue_hires_image.png为高分辨率组织切片图像。
#tissue_lowres_image.png为低分辨率图像,加载和渲染更快,默认时常用
#aligned_tissue_image.jpg为对齐后的组织图像,一般是把组织切片图像和探针阵列对齐后的结果。
#aligned_fiducials.jpg为对齐时使用的参照点
#Fiducials 是 Visium 芯片边缘上的小白点,用来帮助软件自动对齐组织切片和探针阵列。
#detected_tissue_image.jpg为自动识别组织区域后的结果图像,会标出哪些区域被判定为“组织覆盖”,哪些是背景。
#cytassist_image.tiff为扫描仪捕获的组织图像。是后续对齐、生成 aligned_tissue_image 的原始图像来源。
# scalefactors_json.json存放不同图像分辨率与 spot 坐标系统之间的比例系数。
#用于把 barcode 的坐标映射到 hires 或 lowres 图像上。
#tissue_positions.csv(或 .csv.gz)每个 spot 条形码的空间坐标。
#包含条形码 ID 是否落在组织区域 像素坐标(x,y)
#spatial_enrichment.csv 组织区域富集分析相关的信息
二、数据预处理
Show Code
plot1 <- VlnPlot(brain, features = "nCount_Spatial", pt.size = 0.1) + NoLegend()
#pt.size = 0.1:每个点的大小(单个 spot 的数值显示为散点,叠加在小提琴图上)。
plot2 <- SpatialFeaturePlot(brain, features = "nCount_Spatial") + theme(legend.position = "right")
pdf(file="1.nCount_Spatial.pdf",width=20,height=12)
wrap_plots(plot1, plot2)
#前面生成的小提琴图和空间分布图拼到同一画布。
dev.off()
brain <- SCTransform(brain, assay = "Spatial", verbose = FALSE)
#assay = "Spatial":指定输入数据来源于 "Spatial" assay(Visium 数据导入后默认的 assay 名)。
#verbose = FALSE:关闭运行过程的打印信息(让输出更干净)。
#因为空间转录组测序时不同位置的细胞密度不同,因此不同位置表达量的差异不单单表示表达量的高低,
#此时使用LogNormalize()等标准化方法会导致密度等因素的丧失,这里采用SCTransform的方式进行归一化
1.nCount_Spatial.pdf

三、基因表达可视化
Show Code
pdf(file="2.SCTransform.pdf",width=20,height=12)
SpatialFeaturePlot(brain, features = c("Hpca", "Ttr"))
#默认使用brain中 "SCT" assay 的数据,除非你用 assay= 参数指定
#features = c("Hpca", "Ttr"):要展示的基因。
dev.off()
#另一种输出方式
#plot <- SpatialFeaturePlot(brain, features = c("Ttr")) + theme(legend.text = element_text(size = 0), legend.title = element_text(size = 20), legend.key.size = unit(1, "cm"))
#theme修改图例样式:
#legend.text = element_text(size = 0)图例数值文字大小设为 0。
#legend.title = element_text(size = 20)图例标题(Ttr)字号设置为 20。
#legend.key.size = unit(1, "cm")图例色条的大小设为 1 cm 高。
#jpeg(filename = "../output/images/spatial_vignette_ttr.jpg", height = 700, width = 1200, quality = 50)
#quality = 50为JPG 压缩质,范围 0–100,数值越大越清晰但文件越大
#print(plot)
#dev.off()
#SpatialFeaturePlot参数设置
p1 <- SpatialFeaturePlot(brain, features = "Ttr", pt.size.factor = 1)
#pt.size.factor = 1 控制spot大小
p2 <- SpatialFeaturePlot(brain, features = "Ttr", alpha = c(0.1, 1))
#控制 spot 的 透明度。表达值低的 spot透明度 = 0.1,表达值高的 spot透明度 = 1(完全不透明)
pdf(file="3.SCTransform.pdf",width=20,height=12)
p1 + p2
dev.off()
2.SCTransform.pdf

3.SCTransform.pdf

四、降维,聚类和可视化
Show Code
#使用UMAP可视化聚类结果
brain <- RunPCA(brain, assay = "SCT", verbose = FALSE)
brain <- FindNeighbors(brain, reduction = "pca", dims = 1:30)
brain <- FindClusters(brain, verbose = FALSE)
brain <- RunUMAP(brain, reduction = "pca", dims = 1:30)
p1 <- DimPlot(brain, reduction = "umap", label = TRUE)
p2 <- SpatialDimPlot(brain, label = TRUE, label.size = 3)
#使用label = TRUE来在切片图中进行标注
pdf(file="4.UMAP.pdf",width=20,height=12)
p1 + p2
#输出时可能会有好几种报错方式,大概率是Seurat和ggplot2以及patchwork这几个包的问题
#可以更新一下尝试解决
dev.off()
pdf(file="5.CellsByIdentities.pdf",width=20,height=12)
SpatialDimPlot(brain, cells.highlight = CellsByIdentities(object = brain, idents = c(2, 1, 4, 3, 5, 8)), facet.highlight = TRUE, ncol = 3)
#要提取 簇(cluster)编号为 2, 1, 4, 3, 5, 8 的细胞。在空间图上被 高亮显示。
#facet.highlight控制是否单独绘制每个高亮细胞簇的分面图
#SpatialDimPlot(brain, cells.highlight = CellsByIdentities(object = brain, idents = c(2, 1, 4, 3, 5, 8)), facet.highlight = FALSE, ncol = 3, cols.highlight = c("red", "blue", "green", "orange", "purple", "cyan", 'yellow'))
#放在同一张图的写法,注意提供颜色参数
dev.off()
4.UMAP.pdf

5.CellsByIdentities.pdf

五、交互式绘图
Show Code
#交互式窗口
SpatialDimPlot(brain, interactive = TRUE)
#可查看对应spot所属Group
SpatialFeaturePlot(brain, features = "Ttr", interactive = TRUE)
#可交互式调整透明度,点的大小和颜色方案
LinkedDimPlot(brain)
#同时显示UMAP图和组织图像,可以通过点击umap图中区域来高亮组织图像中对应区域,也可通过点击组织区域来高亮对应区域。
SpatialDimPlot

SpatialFeaturePlot
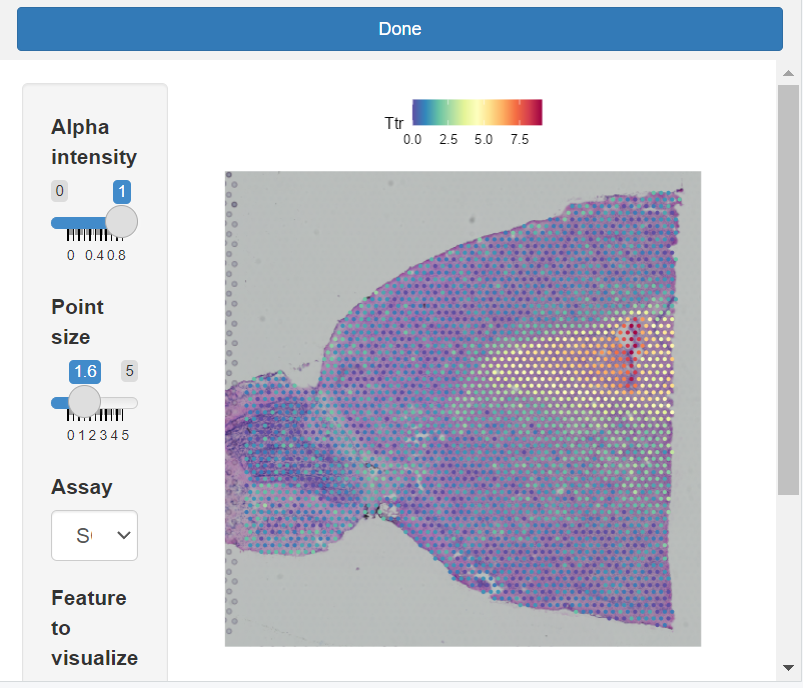
LinkedDimPlot

六、识别空间可变特征
Show Code
#识别空间可变特征
#基于先前的聚类结果进行识别
de_markers <- FindMarkers(brain, ident.1 = 5, ident.2 = 4)
#检测 cluster 5 相对于 cluster 4 显著差异的基因。
SpatialFeaturePlot(object = brain, features = rownames(de_markers)[1:3], alpha = c(0.1, 1), ncol = 3)
#绘制差异分析结果里排名前3的基因在切片上的空间表达分布
#注意默认排序方式是按照p值从小到大排序,如有需要请提前对de_markers进行修改
#alpha = c(0.1, 1)控制 spot 的透明度映射,ncol = 3绘图时一行展示 3 张图
#寻找那些在组织空间分布上不是随机的,而是存在空间模式的基因
brain <- FindSpatiallyVariableFeatures(brain, assay = "SCT", features = VariableFeatures(brain)[1:1000], selection.method = "moransi")
#从对象里取前 1000 个高变基因,使用 Moran’s I 指数 来度量空间自相关。method默认为markvariogram
#跑的慢的话可以试试install.packages('Rfast2')
#VariableFeatures来自SCTransform()的分析结果
#各种报错的可能是各种包版本问题,可以试着把R和R包都更新一波
#结果里面(brain[["SCT"]]@meta.features)只有指定的基因(前1000个)有结果,其余为NA
top.features <- head(SpatiallyVariableFeatures(brain, selection.method = "moransi"), 6)
#对象里提取按 Moran’s I 排序的前六个空间可变基因
pdf(file="6.SpatialFeaturePlot.pdf",width=20,height=12)
SpatialFeaturePlot(brain, features = top.features, ncol = 3, alpha = c(0.1, 1))
#展示得到的基因
dev.off()
6.SpatialFeaturePlot.pdf

七、细分解剖区域
Show Code
##细分解剖区域
cortex <- subset(brain, idents = c(1, 2, 3, 4, 6, 7))
#提取特定Cluster的spot
spatial_coords <- GetTissueCoordinates(cortex)
#用于从空间对象中提取坐标,屏蔽了底层 VisiumV1 和 VisiumV2 类的差异
cortex$imagerow <- spatial_coords[colnames(cortex), "x"] # 注意细胞ID匹配和坐标方向
cortex$imagecol <- spatial_coords[colnames(cortex), "y"]
#将空间坐标添加到 Seurat 对象的元数据中
SpatialDimPlot(cortex,cells.highlight = WhichCells(cortex, expression = imagerow > 5000 & imagecol > 7500))
#可视化筛选区域,辅助边界确定
#以下代码为根据目标区域对解剖区域进行划分
cortex <- subset(cortex, imagerow > 6000 & imagecol > 6000, invert = TRUE)
cortex <- subset(cortex, imagerow > 5000 & imagecol > 7500, invert = TRUE)
#出现大量警告信息只是提示没做合法性验证,不会影响结果
#注意这里追加了invert=TRUE,那么这里的代码不是筛选对应的细胞,而是去掉对应位置的细胞
pdf(file="7.Subset.pdf",width=20,height=12)
p1 <- SpatialDimPlot(cortex, crop = TRUE, label = TRUE)
p2 <- SpatialDimPlot(cortex, crop = FALSE, label = TRUE, pt.size.factor = 1, label.size = 3)
#crop = TRUE:只显示组织区域(切掉空白边缘)。
#crop = FALSE:显示整个载玻片区域(包括背景)。
p1 + p2
dev.off()
7.Subset.pdf

八、与单细胞数据集成
Show Code
##整合单细胞数据
#贴一个从官网找到的数据链接https://www.dropbox.com/s/cuowvm4vrf65pvq/allen_cortex.rds?dl=1
allen_reference <- readRDS("./data/allen_cortex.rds")
#从 RDS 文件中读取 Allen 研究所提供的皮层单细胞 scRNA-seq 数据
#使用的数据不一定是同一块组织的,因为后续会进行匹配,但是推荐同一物种+同一组织+相同测序平台
library(dplyr)
#BiocManager::install('glmGamPoi')
#系统提示安装后能加快一些速度
allen_reference <- SCTransform(allen_reference, ncells = 3000, verbose = FALSE) %>%
RunPCA(verbose = FALSE) %>%
RunUMAP(dims = 1:30)
#SCTransform:使用 sctransform 归一化方法对参考 scRNA-seq 数据进行标准化。
#ncells=3000:对所有细胞做归一化,但只用 3000 个细胞来构建噪声模型。噪声模型用于识别哪些信息是噪声
#save.image("E:/AAA大创/代码注释版/暑假练习/空间转录组/umap.RData")
#跑完及时存储,避免反复跑
cortex <- SCTransform(cortex, assay = "Spatial", verbose = FALSE) %>%
RunPCA(verbose = FALSE)
#对cortex进行分析
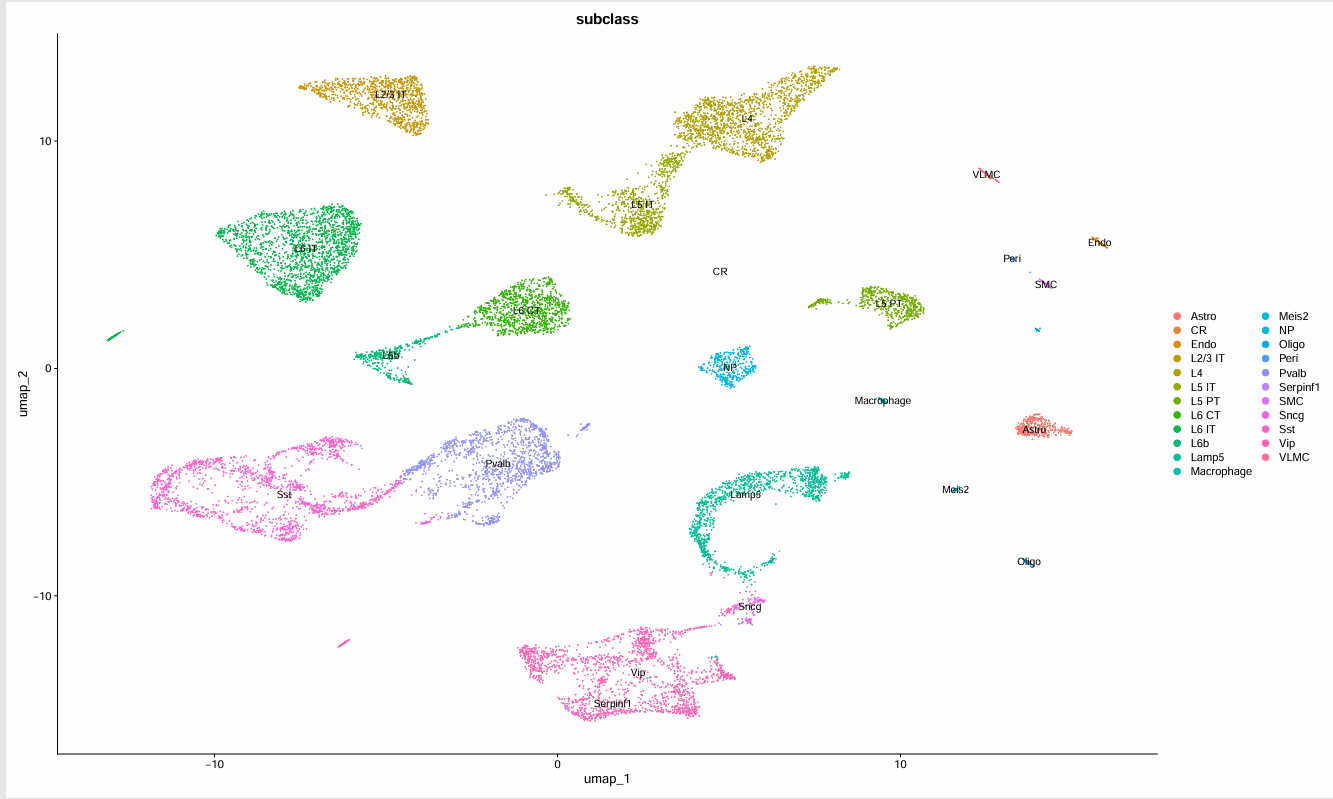
# the annotation is stored in the 'subclass' column of object metadata
pdf(file="8.allen_reference.pdf",width=20,height=12)
DimPlot(allen_reference, group.by = "subclass", label = TRUE)
#按照allen_reference数据中的subclass资料进行分组展示
dev.off()
anchors <- FindTransferAnchors(reference = allen_reference, query = cortex, normalization.method = "SCT")
#作用:在 allen_reference(参考 scRNA-seq 数据)和 cortex(空间转录组数据)之间寻找“锚点(anchors)”
#锚点:就是两组数据中表达模式相似的细胞对,用来对齐和传递信息。
#normalization.method = "SCT":表示使用 SCTransform 标准化方式来进行匹配。
prediction.assay = TRUE
#默认情况下,TransferData() 会把参考数据的标签直接写入 query 对象的 meta.data
#但是如果你设置 prediction.assay = TRUE,它会在 query 对象中新建一个名为 "predictions"的 Assay,存储概率矩阵:
#表示该细胞属于对应标签的概率(介于 0~1)。
predictions.assay <- TransferData(anchorset = anchors, refdata = allen_reference$subclass, prediction.assay = TRUE,
weight.reduction = cortex[["pca"]], dims = 1:30)
#作用:把参考数据中的注释(subclass,也就是细胞亚类)转移到 cortex 上。
#anchors: 上一步找好的锚点。
#refdata = allen_reference$subclass: 把参考数据中 subclass 注释当作要转移的标签。
#prediction.assay = TRUE: 结果存放在一个新的 assay 里(名为 "predictions")。
#weight.reduction = cortex[["pca"]]: 在 PCA 降维空间中做标签转移。
#dims = 1:30: 使用前 30 个主成分。
cortex[["predictions"]] <- predictions.assay
DefaultAssay(cortex) <- "predictions"
#把预测结果存到 cortex 里,并且设置 "predictions" 作为默认 assay(后续可直接调用预测分数)。
pdf(file="9.cortex_predictioins.pdf",width=20,height=12)
SpatialFeaturePlot(cortex, features = c("L2/3 IT", "L4"), pt.size.factor = 1.6, ncol = 2, crop = TRUE)
#展示L2/3 IT、L4两组,同时使用crop裁剪掉多余的空白区域。
dev.off()
cortex <- FindSpatiallyVariableFeatures(cortex, assay = "predictions", selection.method = "moransi",
features = rownames(cortex), r.metric = 5, slot = "data")
#在 cortex Seurat 对象里,寻找空间上变化显著的基因/特征。这里指定使用Moran’s I来选择空间可变特征。
#使用的是存放在 predictions assay 里的数据
#features指定要测试的特征,即"Vip""Lamp5""Sst""Sncg"等
#r.metric规定局部邻域多少范围内的点纳入统计
#slot = "data"使用 assay 中的 data 层而不是count或者scale.data层,虽然是概率矩阵,但是这两层仍然存在
top.clusters <- head(SpatiallyVariableFeatures(cortex, selection.method = "moransi"), 4)
#空间可变特征里取前4个最显著的特征
pdf(file="10.predictioins_SpatialPlot.pdf",width=20,height=12)
SpatialFeaturePlot(object = cortex, features = top.clusters, ncol = 2)
dev.off()
#展示选出的前4个可变特征的空间分布
pdf(file="11.predictioins_SpatialFeaturePlot.pdf",width=20,height=12)
SpatialFeaturePlot(cortex, features = c("Astro", "L2/3 IT", "L4", "L5 PT", "L5 IT", "L6 CT", "L6 IT",
"L6b", "Oligo"), pt.size.factor = 1, ncol = 2, crop = FALSE, alpha = c(0.1, 1))
#展示指定类型的空间分布
dev.off()
8.allen_reference.pdf
9.cortex_predictioins.pdf

10.predictioins_SpatialPlot.pdf
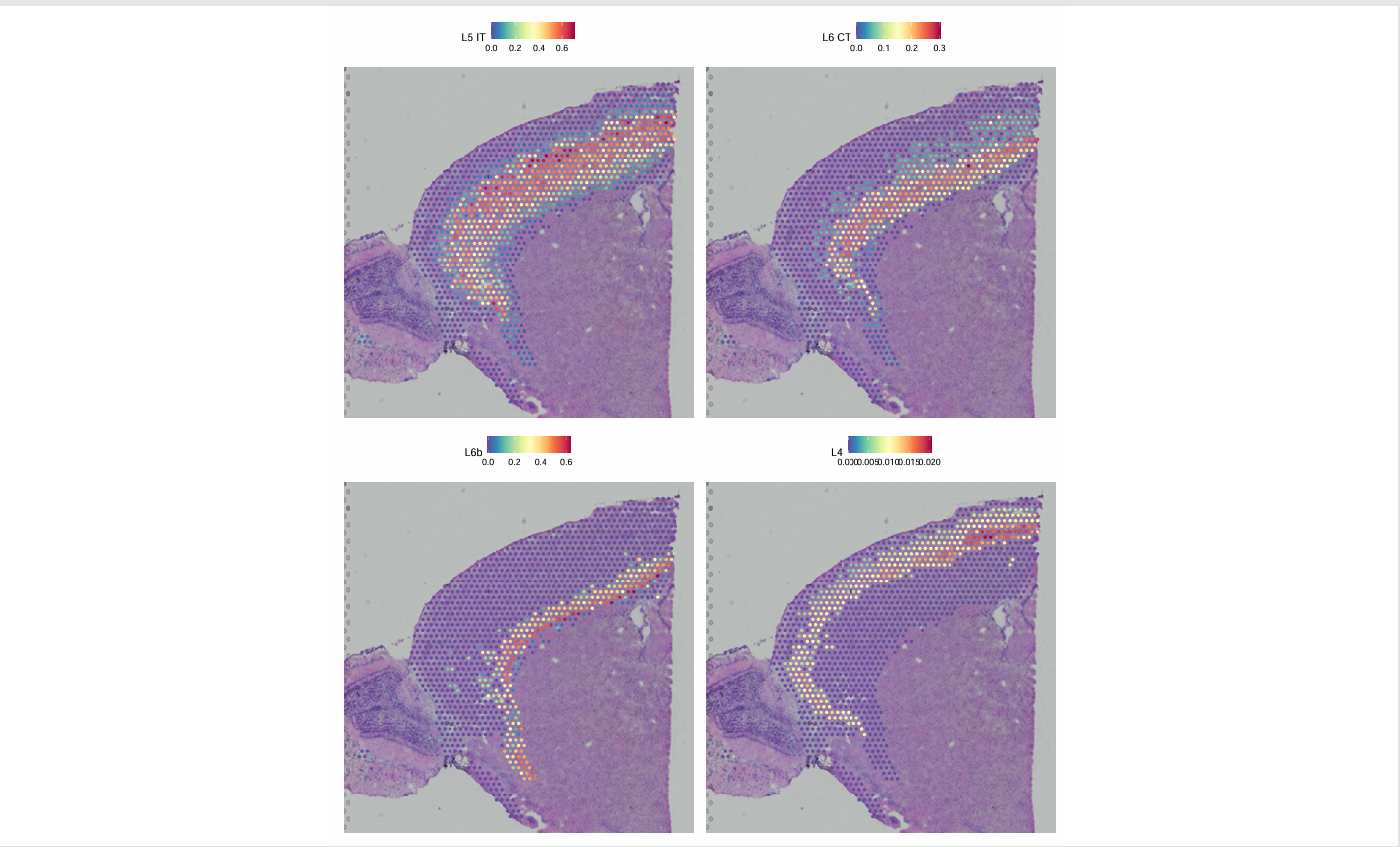
11.predictioins_SpatialFeaturePlot.pdf

九、在Seurat中处理多个切片
Show Code
##Seurat处理多个切片
brain2 <- LoadData("stxBrain", type = "posterior1")
#数据集包含另一个与大脑另一半相对应的切片。
brain2 <- SCTransform(brain2, assay = "Spatial", verbose = FALSE)
#合并两个切片数据
brain.merge <- merge(brain, brain2)
DefaultAssay(brain.merge) <- "SCT"
VariableFeatures(brain.merge) <- c(VariableFeatures(brain), VariableFeatures(brain2))
brain.merge <- RunPCA(brain.merge, verbose = FALSE)
brain.merge <- FindNeighbors(brain.merge, dims = 1:30)
brain.merge <- FindClusters(brain.merge, verbose = FALSE)
brain.merge <- RunUMAP(brain.merge, dims = 1:30)
DimPlot(brain.merge, reduction = "umap", group.by = c("ident", "orig.ident"))
#ident:按照聚类结果上色
#orig.ident:按照样本来源上色
pdf(file="12.merge_SpatialDimPlot.pdf",width=20,height=12)
SpatialDimPlot(brain.merge)
dev.off()
pdf(file="13.merge_SpatialFeaturePlot.pdf",width=20,height=12)
SpatialFeaturePlot(brain.merge, features = c("Hpca", "Plp1"))
#SpatialDimPlot()和 SpatialFeaturePlot()的默认排版是将不同切片绘制为列,将不同分组/要素绘制为行。
dev.off()
12.merge_SpatialDimPlot.pdf

13.merge_SpatialFeaturePlot.pdf

???
数据真大,我E盘又红了
尤其在运行
allen_reference <- SCTransform(allen_reference, ncells = 3000, verbose = FALSE) %>%
RunPCA(verbose = FALSE) %>%
RunUMAP(dims = 1:30)
的时候,最好把后台清一清,否则电脑容易卡死,还跑不出来结果



 浙公网安备 33010602011771号
浙公网安备 33010602011771号