
fastqc安装及使用
fastqc是高通量序列(fa/fq)质量控制(QC)分析工具。它可以快速地对测序数据进行质量评估,得到多个测序数据的质量参数,让我们对测序数据质量有个初步的认识,从而判断后续的质控如何进行。
一、安装
conda install -c bioconda fastqc二、语法
fastqc seqfile1 seqfile2 .. seqfileN
fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam] [-c contaminant file] seqfile1 .. seqfileN三、使用
fastqc -o <output dir> -t 16 <in.R1.fq.gz> <in.R2.fq.gz>四、参数
-o --outdir FastQC生成的报告文件的储存路径,生成的报告的文件名是根据输入来定的
-f --format 指定输入文件的格式
--extract 生成的报告默认会打包成1个压缩文件,使用这个参数是让程序不打包
-t --threads 选择程序运行的线程数,每个线程会占用250MB内存,越多越快咯
--min_length 设置序列的最小长度,≥最长read的长度
-c --contaminants 污染物选项,输入的是一个文件,格式是Name [Tab] Sequence,里面是可能的污染序列,如果有这个选项,FastQC会在计算时候评估污染的情况,并在统计的时候进行分析,一般用不到
-a --adapters 也是输入一个文件,文件的格式Name [Tab] Sequence,储存的是测序的adpater序列信息,如果不输入,目前版本的FastQC就按照通用引物来评估序列时候有adapter的残留
-q --quiet 安静运行模式,一般不选这个选项的时候,程序会实时报告运行的状况五、脚本批处理
PS: 处理重测序数据时非常需要,运行脚本前别忘了切换至工作目录(存放批量fastq文件的目录下),同时需要创建输出文件夹FASTQC存储输出文件
具体可参考如下脚本:
#!/bin/bash
#program:
# Checking sequencing reads quality with FASTQC
#2022/9/16
for id in *fq
do
echo $id
fastqc -o /home/liuxin/output/FASTQC -t 16 --extract $id
done具体操作如下:
vim ngs_fastqc.sh #添加脚本内容并修改处理文件格式fq/fa或者fastq/fasta,并调整输出路径即可
chmod u+x ngs_fastqc.sh
bash ngs_fastqc.sh运行情况如下:
六、结果查看
生成的HTML文件可本地化后查看,其中包含分门别类的统计信息,如基本统计信息、GC含量、碱基位点质量等。查看HTML结果如下:
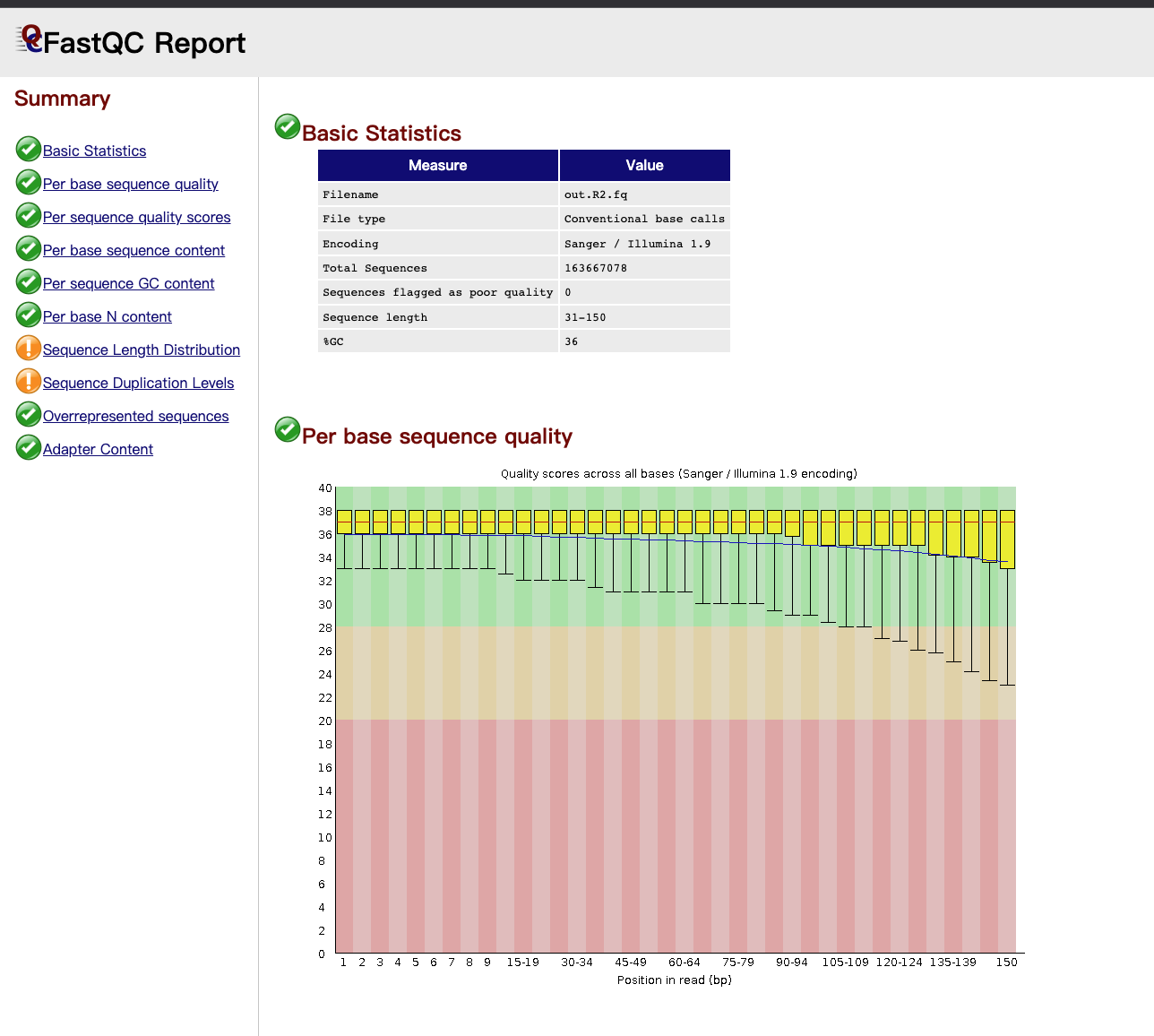
PS:后续有时间再对结果报告深入研究。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号