【统计学代码包计划】倾向评分匹配
Matchit包只是一个匹配的工具,具体的距离(distance)算法以及匹配方法(method)可以自由更换
首先选择想要匹配的因素进行匹配,距离计算方法通常用logistic回归法
参数解释:(cite:https://zhuanlan.zhihu.com/p/596876245)
caliper:卡钳值,也就是配对标准,两组的概率值(PS)差距在这个标准内才会配对。这里的卡钳值是PS标准差的倍数,默认是不设置卡钳值。还有一个std.caliper参数,默认是TRUE,如果设置FALSE,你设置的卡钳值就直接是PS的倍数。replace:能否重复匹配,默认是FALSE,意思是假如干预组的1号匹配到了对照组的A,那A就不能再和其他的干预组进行匹配了。ratio:设置匹配比例,干预组:对照组到底是1比几,默认为1:1。ratio=2即是干预组:对照组是1:2。所以一般要求数据的对照组数量多于干预组才行。如果对照组比干预组多出很多,完全可以设置1:n进行匹配,这样还能损失更少的样本信息,但是一般也不会超过1:4。reestimate:如果是TRUE,丢掉没匹配上的样本,PS会使用剩下的样本重新计算PS,如果是FALSE或者不写就不会重新计算PS。
匹配算法有下面的:(cite:https://zhuanlan.zhihu.com/p/596876245)
- 默认的匹配方法是最近邻匹配nearest,其他方法还有
- "exact" (exact matching),
- "full" (optimal full matching),
- "optimal" (optimal pair matching),
- "cardinality"(cardinality and template matching),
- "subclass" (subclassification),
- "genetic" (genetic matching),
- "cem" (coarsened exact matching)
m.out <- matchit(diagnosis==2~age+CCT,
data = tmp,
distance = "logit",
ratio=5, #1:5匹配
method='nearest')
m.out#输出看看匹配的结果,有多少匹配上了多少没有
会输出这个

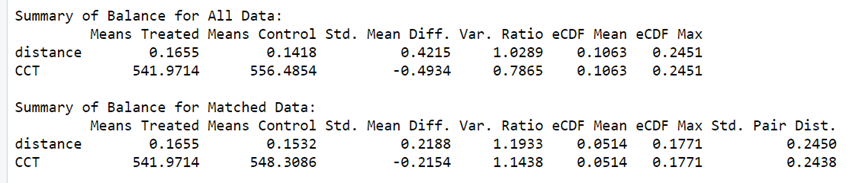
用matched/(1+5)就知道有多少个病例组匹配上了,然后检测匹配前后的SMD,这个文章里需要汇报
summary(m.out,standardize = TRUE)
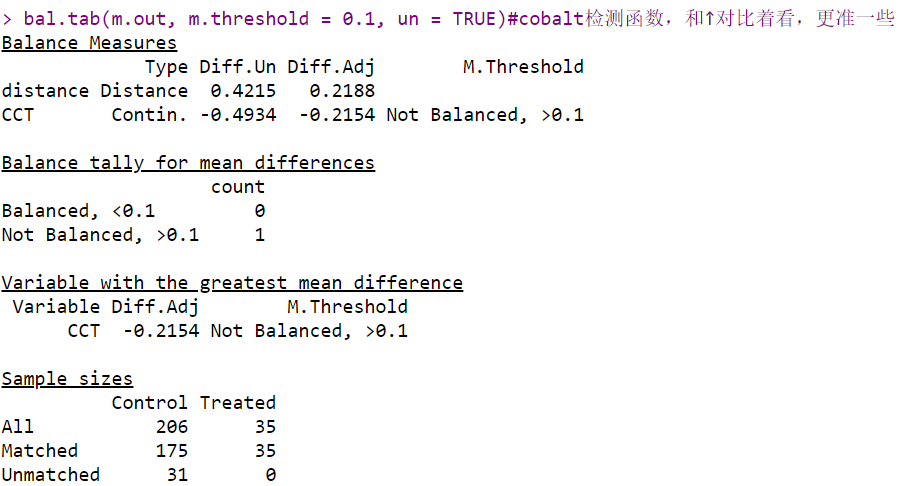
#matchit自带的SMD检测函数,据说不是很准 bal.tab(m.out, m.threshold = 0.1, un = TRUE)
#cobalt检测函数,和↑对比着看,更准一些
第一种方法结果:
第二种:
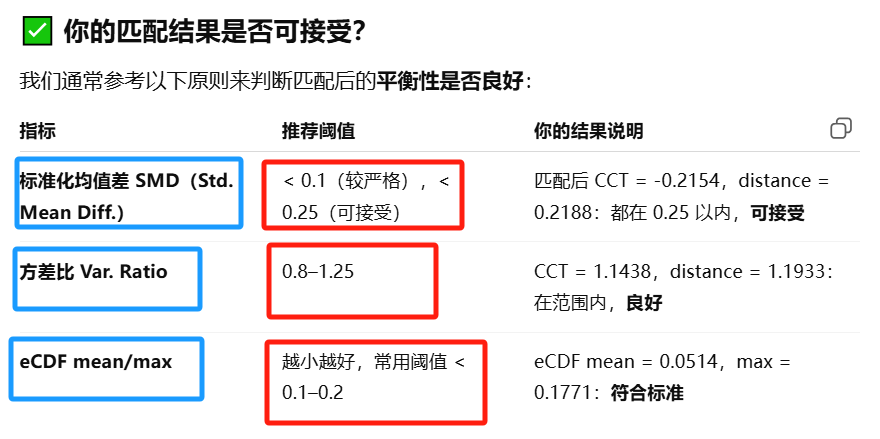
怎么解读,GPT告诉你:
然后可以看看自己匹配完了两组还有没有差异
mdata <- match.data(m.out)#把匹配好的存在一个新的数据结构里
#画一个基线表来检测匹配的记过
baseline<-descrTable( diagnosis~.,
data = mdata,
sd.type = 2,
q.type = c(2, 2), #四分位间距显示形式
method = NA,
hide.no = "0",
digits = 2,show.p.overall = T)
summary(baseline)
export2word(baseline, file='baseline after match.docx')

 浙公网安备 33010602011771号
浙公网安备 33010602011771号