构建聚合物分子模型的示例
1 引言
在分子动力学模拟中,最容易忘记的就是命令,但是最容易查到的也是命令,为了让我之后能更加连贯的工作(摸鱼),也希望能给看官一个参考。
2 构建分子链
这里强推sob老师的计算化学论坛。方法来源也是:基于OpenBabel批量产生特定基团以任意方式接到苯上的结构的方法 - 分子模拟 (Molecular Modeling) - 计算化学公社 (keinsci.com)
首先要构建的就是聚合物的分子链,这里采用的是obabel软件的gen3d功能,可以将smiles字符串转化为任意格式的三维结构,虽然具体的构象仍需考虑,但是作为开始,可以说是万能之选。(关于什么是smiles字符串,我建议你参考:简化分子线性输入规范 - 维基百科 ,不要觉得麻烦,事实上这很容易理解。)
第一步构建单体,因为聚合物由于聚合度不同,我们所使用的smiles字符串也不同,所以为了简便,我们利用smiles字符串的优势,只构建了单体,用程序(或者手动)将单体放到一行即可实现单体的“聚合”。(这里请注意分子在中间和端时的写法可能稍微有区别)
(note:这个构建方式对于线性聚合物比较友好,如果是体型结构,py程序需要大改下,可能得递归)
我这里是写了一个含有苯并咪唑杂环的芳香族聚酰胺分子(杂环芳纶):
分子结构:
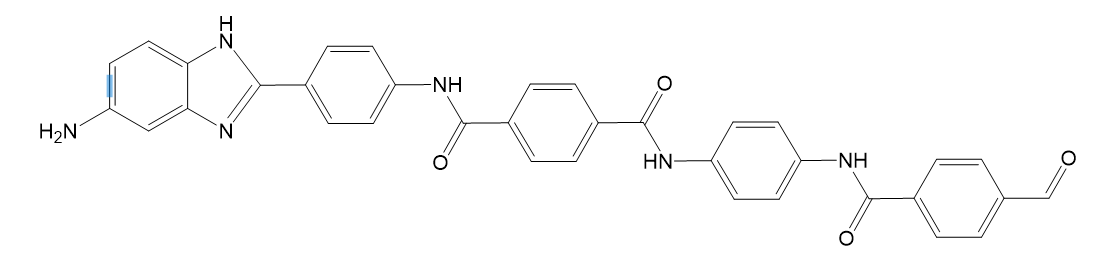
端的芳纶(poly_begin):
O=Cc(cc1)ccc1C(=O)NCc(cc1)ccc1C(=O)Nc(cc1)ccc1NC(=O)c(cc1)ccc1C(=O)Nc(cc1)ccc1-c2nc(c1)c([nH]2)ccc1N
中间(poly_mid):
C(=O)c(cc1)ccc1C(=O)NCc(cc1)ccc1C(=O)Nc(cc1)ccc1NC(=O)c(cc1)ccc1C(=O)Nc(cc1)ccc1-c2nc(c1)c([nH]2)ccc1N
#二者区别仅在于最开头的碳氧双键的写法,后者作为中间,前者为开端。
#伪代码如下:
num = 4 #聚合度
filename_wri = f'./sp{num}.smi'
with open(filename_wri, 'w') as file:
file.write(poly_begin)
for i in range(0, num-1):
file.write(poly_mid)之后使用obabel转化smiles字符串为三维结构,这里采用MMFF94力场进行能量最小化。
tips:一般分子越长越复杂等待时间会越长,同时如果你的分子链聚合度过高,需要考虑你的电脑的内存是否可以承受,这里建议使用非GUI版本,GUI版本容易卡住
obabel shili.smi -O out.pdb --gen3d -m --minimize --ff MMFF94 # -minimize 能量最小化 -O 后面的文件格式可以调,我用的一般是mol2文件格式3 计算原子电荷(charge)
这里如何想省事,只是进行分子动力学模拟,可以直接使用sobtop,用MMFF94分配原子电荷。
我使用的是multiwfn的Resp, (懒人脚本参考:计算RESP原子电荷的超级懒人脚本(一行命令就算出结果) - 分子模拟 (Molecular Modeling) - 计算化学公社 (keinsci.com))
计算出的.chg文件不能直接使用,需要手动匹配一下,同时匹配的注意事项参考下文
Hint:关于给聚合物用RESP原子电荷
具体方法参考:Sobtop (sobereva.com)和 RESP拟合静电势电荷的原理以及在Multiwfn中的计算 - 波函数分析与Multiwfn (Wavefunction Analysis & Multiwfn) - 计算化学公社 (keinsci.com)
对分子进行原子性约束和等价,,,,
3 单分子模型计算(如键解离能计算)
有了分子模型之后,如果想对分子进行键能,静电势等方面计算,需要首先使用mopac进行半经验方法的初步几何优化(PM6就可以了),然后再用Orca分别计算键解离能(这里直接按照orca官方的orca_maual手册即可编写)
4 构建异形聚合物模型
有了单分子,使用Packmol即可任意进行排布,这里推荐sob的一些脚本示例帮助理解,熟能生巧。
本文来自博客园,作者:{lufenghao},转载请注明原文链接:https://www.cnblogs.com/lfhnb/p/18202555

 浙公网安备 33010602011771号
浙公网安备 33010602011771号