

原文:Chip-seq上游分析流程 (附代码) | 新手可入(点击进入)
本期图形
写在前面
最近做了一个Chip-seq的上游分析教程(其实,也不是“做的”,就是学习一下,都是基于以下哪些优秀的教程,综合一下)。顺便跑了番茄Chip-seq数据,我们学习时尽量把数据带进去。这样便于验证自己所学的代码是否错误,不能运行。
在web中的教程,可能一些教程自己运行时会有报错。因此,自己上机操作是非常有必要的。
此外,我们教程中,也使用了Singularity容器(Linux软件安装, singularity容器中安装软件(我们计划花时间,将日常用到的软件进行封装)),以及制作了对软件进行打包,你可以直接拿过去,上机即可操作。
注: 我们已在社群中的同学,可以直接在对应网盘中获得本期资源数据代码。
$ tree ../ChipSeq_PRJNA1124576/
../ChipSeq_PRJNA1124576/
├── 00_Raw_data
│ ├── 01.run.down.PRJNA1124576.sh
│ ├── SRR33408969_1.fastq.gz
│ ├── SRR33408969_2.fastq.gz
│ ├── SRR33408970_1.fastq.gz
│ ├── SRR33408970_2.fastq.gz
│ ├── SRR33408971_1.fastq.gz
│ ├── SRR33408971_2.fastq.gz
│ ├── SRR33408972_1.fastq.gz
│ ├── SRR33408972_2.fastq.gz
│ ├── SRR33408973_1.fastq.gz
│ ├── SRR33408973_2.fastq.gz
│ ├── SRR33408974_1.fastq.gz
│ ├── SRR33408974_2.fastq.gz
│ ├── data.list.txt
│ └── nohup.out
├── 01_Clean_Chipsep
│ ├── SRR33408969_1.clean.fastq.gz
│ ├── SRR33408969_2.clean.fastq.gz
│ ├── SRR33408970_1.clean.fastq.gz
│ ├── SRR33408970_2.clean.fastq.gz
│ ├── SRR33408971_1.clean.fastq.gz
│ ├── SRR33408971_2.clean.fastq.gz
│ ├── SRR33408972_1.clean.fastq.gz
│ ├── SRR33408972_2.clean.fastq.gz
│ ├── SRR33408973_1.clean.fastq.gz
│ ├── SRR33408973_2.clean.fastq.gz
│ ├── SRR33408974_1.clean.fastq.gz
│ ├── SRR33408974_2.clean.fastq.gz
│ └── html
│ ├── SRR33408969.html
│ ├── SRR33408969.json
│ ├── SRR33408970.html
│ ├── SRR33408970.json
│ ├── SRR33408971.html
│ ├── SRR33408971.json
│ ├── SRR33408972.html
│ ├── SRR33408972.json
│ ├── SRR33408973.html
│ ├── SRR33408973.json
│ ├── SRR33408974.html
│ └── SRR33408974.json
├── 02_Geneome_ref
│ ├── Tomato_index_bowtie2
│ │ ├── Sl.1.bt2
│ │ ├── Sl.2.bt2
│ │ ├── Sl.3.bt2
│ │ ├── Sl.4.bt2
│ │ ├── Sl.rev.1.bt2
│ │ └── Sl.rev.2.bt2
│ └── Tomato_ref_4.0
│ ├── ITAG4.0_CDS.fasta
│ ├── ITAG4.0_cDNA.fasta
│ ├── ITAG4.0_gene_models.bed
│ ├── ITAG4.0_gene_models.gff
│ ├── ITAG4.0_gene_models.gtf
│ ├── ITAG4.0_proteins.fasta
│ ├── S_lycopersicum_chromosomes.4.00.fa
│ └── genome_size.py
├── 03-04.run.sh
├── 03_mapped
│ ├── 0301_rmdup
│ │ ├── SRR33408969_rmdup_flagstat.txt
│ │ ├── SRR33408970_rmdup_flagstat.txt
│ │ ├── SRR33408971_rmdup_flagstat.txt
│ │ ├── SRR33408972_rmdup_flagstat.txt
│ │ ├── SRR33408973_rmdup_flagstat.txt
│ │ └── SRR33408974_rmdup_flagstat.txt
│ ├── SRR33408969.bam
│ ├── SRR33408969.sam
│ ├── SRR33408969_flagstat.txt
│ ├── SRR33408970.bam
│ ├── SRR33408970.sam
│ ├── SRR33408970_flagstat.txt
│ ├── SRR33408971.bam
│ ├── SRR33408971.sam
│ ├── SRR33408971_flagstat.txt
│ ├── SRR33408972.bam
│ ├── SRR33408972.sam
│ ├── SRR33408972_flagstat.txt
│ ├── SRR33408973.bam
│ ├── SRR33408973.sam
│ ├── SRR33408973_flagstat.txt
│ ├── SRR33408974.bam
│ ├── SRR33408974.sam
│ └── SRR33408974_flagstat.txt
├── 04_Rename
│ ├── OEJMJ4_HA_rep1.bam
│ ├── OEJMJ4_HA_rep1_sort.bam
│ ├── OEJMJ4_HA_rep2.bam
│ ├── OEJMJ4_HA_rep2_sort.bam
│ ├── OEJMJ4_HA_rep3.bam
│ ├── OEJMJ4_HA_rep3_sort.bam
│ ├── OEJMJ4_Input_rep1.bam
│ ├── OEJMJ4_Input_rep1_sort.bam
│ ├── OEJMJ4_Input_rep2.bam
│ ├── OEJMJ4_Input_rep2_sort.bam
│ ├── OEJMJ4_Input_rep3.bam
│ ├── OEJMJ4_Input_rep3_sort.bam
│ ├── bedtools_intersect.log
│ ├── id_norep.txt
│ ├── intersect
│ │ ├── OEJMJ4_HA_intersect.bam
│ │ ├── OEJMJ4_HA_intersect.bam.bai
│ │ └── OEJMJ4_Input_intersect.bam
│ ├── macs2
│ │ ├── OEJMJ4_HA_intersect_model.pdf
│ │ ├── OEJMJ4_HA_intersect_model.r
│ │ ├── OEJMJ4_HA_intersect_peaks.narrowPeak
│ │ ├── OEJMJ4_HA_intersect_peaks.xls
│ │ └── OEJMJ4_HA_intersect_summits.bed
│ └── rename.txt
├── 05_bamCoverage
│ └── OEJMJ4_HA_intersect.bw
├── 06_Peak_heatmap
│ ├── HA_TSS.png
│ ├── HA_TSS02.pdf
│ └── HA_TSS_matrix.gz
├── Script
│ ├── 01.fastp.run.sh
│ ├── 02.bowtie2_index.sh
│ ├── 03.bowtie2_mapped.sh
│ ├── 04.samtools_markdup.sh
│ ├── 05.rename.sh
│ ├── 06_bedtools_intersect.sh
│ ├── 07.macs2_callpeak.sh
│ └── 08.bam_To_bw.sh软件部分,我们仅制作了几个,后续我们持续更新。
$ tree ../SingularityLib/
../SingularityLib/
├── CNCI.sif
├── CPC2.sif
├── MACS2.sif
├── bcftools.sif
├── bedtools.sif
├── blast.sif
├── bowtie2.sif
├── diamond.sif
├── fastp.sif
├── hifiasm.sif
├── hisat2.sif
├── jellyfish.sif
├── metilene.sif
├── pfastq-dump.sif
└── sratoolkit.sif**注:**本教程相对较长,结合自己的时间进行学习即可。我们也将学习参考的教程进行引用。大家也可以直接查看参考教程进行学习。
一、参考
1.1 Chip-seq测序原理
ChIP-seq(Chromatin Immunoprecipitation Sequencing,染色质免疫沉淀测序)的核心是定位细胞内特定蛋白质(如转录因子、组蛋白修饰)结合的DNA区域,从而揭示蛋白质如何调控基因表达或染色质状态。
核心原理:“特异性捕获+测序定位”
蛋白质与DNA在细胞内通常处于动态结合状态,ChIP-seq通过以下逻辑实现结合位点的鉴定:
- 交联固定:用甲醛处理细胞,使体内结合的“蛋白质-DNA复合物”共价交联,冻结其相互作用状态(避免后续操作中复合物解离);
- 破碎染色质:用超声将染色质打断为100-500 bp的短片段(片段大小直接影响后续分辨率);
- 免疫沉淀(IP):加入针对目标蛋白质的特异性抗体,通过抗原-抗体反应捕获“目标蛋白-DNA复合物”(这是ChIP-seq的关键瓶颈,抗体特异性直接决定结果真实性);
- 解交联与纯化:用蛋白酶K消化蛋白质,释放结合的DNA片段,再纯化DNA;
- 测序分析:对纯化的DNA片段构建测序文库并测序,将测序 reads 比对到参考基因组,富集的区域即为“目标蛋白质的结合位点”。
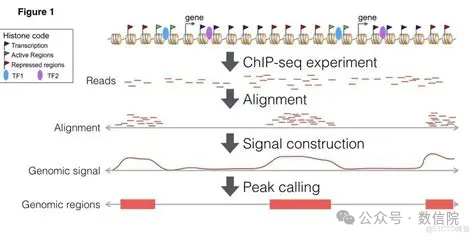
数据分析核心步骤
- 数据质控:用FastQC过滤低质量reads、去除接头序列(如Trimmomatic);
- 基因组比对:用BWA或Bowtie2将clean reads比对到参考基因组,去除重复reads(如Picard);
- 峰 calling(找结合位点):用MACS3(最常用工具)通过“处理组vs输入对照组(Input DNA)”的信号差异,识别显著富集的“峰”(Peak),峰的位置即蛋白质结合的DNA区域;
- 峰注释:用ChIPseeker等工具将峰关联到基因组功能区域(如启动子、增强子、内含子),明确结合位点调控的靶基因;
- 功能分析:对靶基因进行GO功能注释、KEGG通路富集分析,揭示目标蛋白的生物学功能;
- 可视化:用IGV(Integrative Genomics Viewer)展示峰在基因组上的位置,用Heatmap或Motif分析(如HOMER)寻找峰序列中富集的转录因子结合基序(Motif)。
软件安装:
- 使用
mamba进行安装
mamba install -c bioconda -y fastp bowtie2 samtools bedtools \
deeptools macs2使用mamba软件,可能出现deeptools macs2所需程序与本地程序有冲突,如python版本等。
- 使用Singularity容器安装
bowtie2
网址:
https://sourceforge.net/projects/bowtie-bio/
创建bowtie2.sif
$ singularity build --sandbox bowtie2 ubuntu-latest_latest.sif
INFO: Starting build...
INFO: Verifying bootstrap image ubuntu-latest_latest.sif
WARNING: integrity: signature not found for object group 1
WARNING: Bootstrap image could not be verified, but build will continue.
INFO: Creating sandbox directory...
INFO: Build complete: bowtie2下载最新版本
wget https://sourceforge.net/projects/bowtie-bio/files/bowtie2/2.5.4/bowtie2-2.5.4-sra-linux-x86_64.zip
unzip bowtie2-2.5.4-sra-linux-x86_64.zip
cd bowtie2-2.5.4-sra-linux-x86_64添加环境变量
vim bowtie2/environment
##输入路径
PATH=$PATH:/opt/bowtie2-2.5.4-sra-linux-x86_64打包软件
Singularity build -f bowtie2.sif bowtie2/fastp安装
- 软件网址
https://github.com/OpenGene/fastp
- 软件下载
https://github.com/OpenGene/fastp/releases
wget https://github.com/OpenGene/fastp/archive/refs/tags/v1.0.1.tar.gz- 解压与安装
tar -zxvf fastp-1.0.1.tar.gz
##
cd fastp-1.0.1
make -j- 添加环境变量
vim ioinfR/environment
## 添加路径
PATH=$PATH:/opt/fastp-1.0.1
bedtools安装
apt-get update && apt install -y --no-install-recommends build-essential
apt-get install -y zlib1g-dev
#
apt-get install -y libbz2-dev
apt-get install -y liblzma-dev- 下载
https://github.com/arq5x/bedtools2?tab=readme-ov-filehttps://github.com/arq5x/bedtools2/releases/tag/v2.31.1
- 解压与安装
tar -zxvf bedtools-2.31.1.tar.gz
cd bedtools2/
## 安装/编译
make- 使用教程
https://bedtools.readthedocs.io/en/latest/
samtools
查看安装命令
$ cat README
##------------
The typical simple case of building Samtools using the HTSlib bundled within
this Samtools release tarball is done as follows:
cd .../samtools-1.22 # Within the unpacked release directory
./configure
make- 下载软件
https://github.com/samtools/samtools#
- 解压与安装
tar -xvjf samtools-1.22.tar.bz2
cd samtools-1.22
## 安装
./configure
make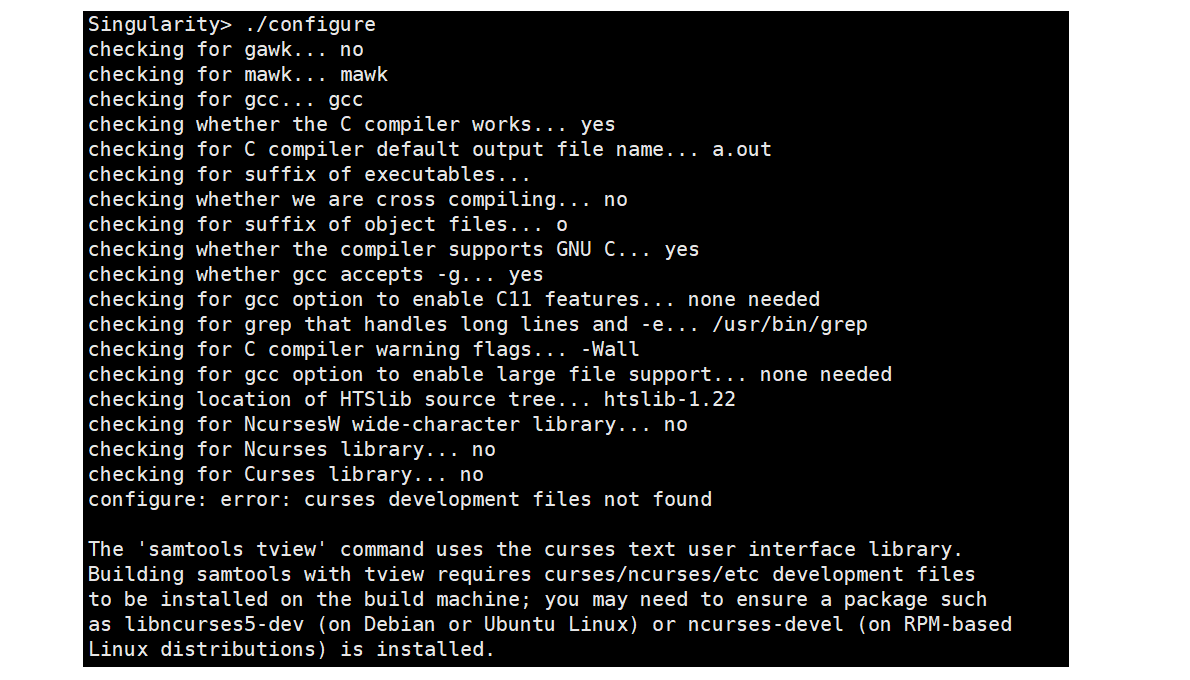
- 添加环境变量
vim ioinfR/environment
##输入路径
PATH=$PATH:/opt/samtools-1.22
下载最新版本。
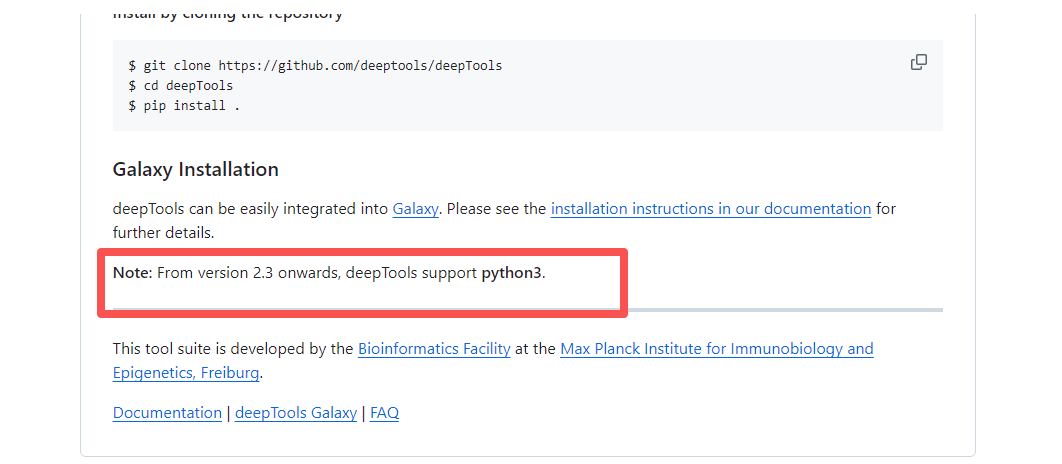
此软件需要python版本为python3
apt-get update && apt install -y --no-install-recommends build-essential \
python3 python3-pip python3-dev软件安装
$ git clone https://github.com/deeptools/deepTools
$ cd deepTools
$ pip install .
MACS2
软件网址:
https://pypi.org/project/MACS2/#files
创建MACS2沙箱
$ singularity build --sandbox MACS2 ubuntu-latest_latest.sif
## 下载软件
wget https://files.pythonhosted.org/packages/1f/a7/973a7867efc40ff87d040f3adac29a26fcde76edbf10ed80733c1caafbf6/MACS2-2.2.9.1.tar.gz
## 解压与安装
tar -zxvf MACS2-2.2.9.1.tar.gz
cd MACS2-2.2.9.1
#此软件需要python版本为python3
apt-get update && apt install -y --no-install-recommends build-essential \
python3 python3-pip python3-dev## 安装
pip install Numpy ## 所需python 模块
## 安装
pip install .给
python创建软链接指向 Python3
ln -s /usr/bin/python3 /usr/bin/python这样
/usr/bin/env python就能找到 Python3 了。
## 路径
vim MACS2/environment
##
PATH=$PATH:/opt/MACS2-2.2.9.1/bin
二、数据下载
2.1 参考数据-Tomato CHIP-seq
- 数据来自NCBI,项目编号:PRJNA1124576
网址:
https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1124576/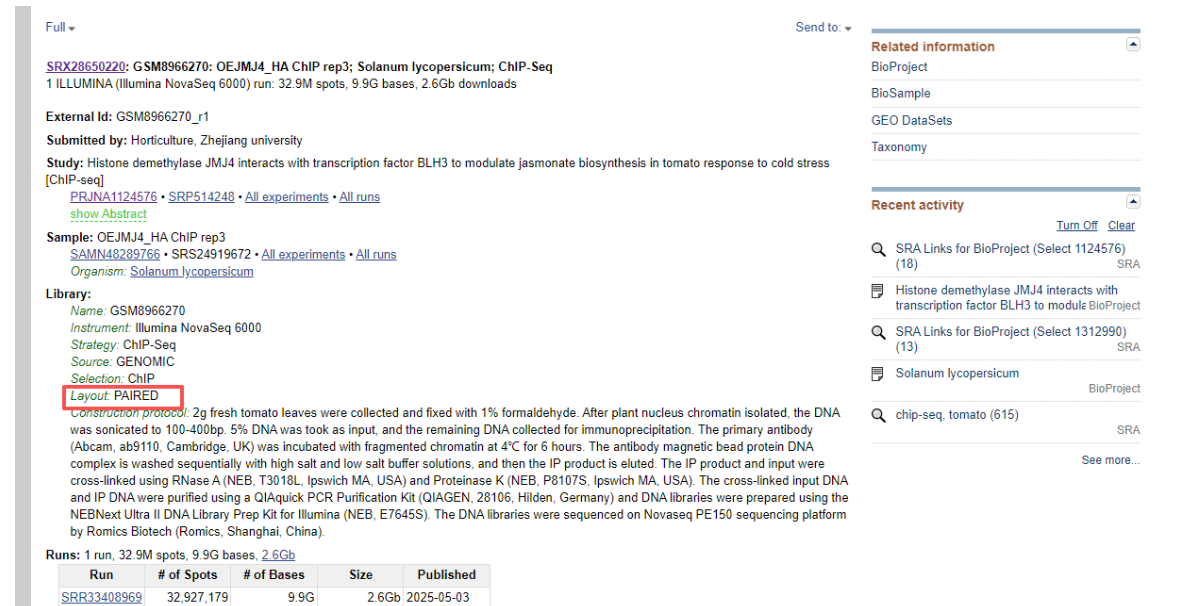
- 数据下载脚本
使用kingfisher快速下载。
kingfisher get -p PRJNA1124576 -m aws-http prefetch aws-cp gcp-cp ena-ascp ena-ftp -t 30 -f fastq.gz --check-md5sums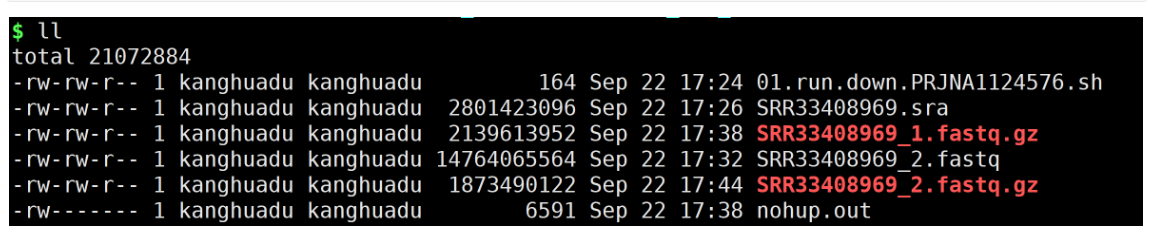
2.2 数据质控
我们只是为了记录操作流程,因此,只选择6个fq数据进行后续的分析。
## data SRA
SRR33408969 SRR33408970 SRR33408971 SRR33408972 SRR33408973 SRR33408974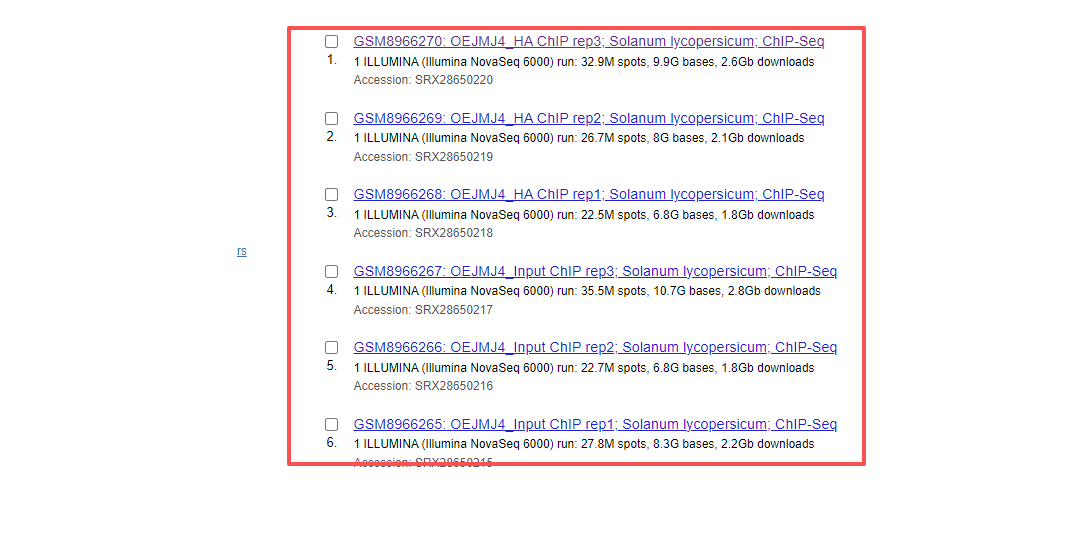
Code
mkdir 01_Clean_Chipsep**01.fastp.run.sh**
#!/bin/bash
## 在后面的程序中直接修改“Raw"和”Clean"的文件位置即可
Raw="/home/user/ChipSeq_PRJNA1124576/00_Raw_data"
Clean="/home/user/ChipSeq_PRJNA1124576/01_Clean_Chipsep"
Html="/home/user/ChipSeq_PRJNA1124576/01_Clean_Chipsep/html"
##
cat ${Raw}/data.list.txt | awk '{print $1}' | while read id;
do
time singularity exec ~/SingularityLib/fastp.sif fastp -w 16 \
-i ${Raw}/${id}_1.fastq.gz -o ${Clean}/${id}_1.clean.fastq.gz \
-I {Raw}/${id}_2.fastq.gz -O ${Clean}/${id}_2.clean.fastq.gz \
-j ${Html}/${id}.json \
-h ${Html}/${id}.html
done注意:
在添加路径时,请不要使用~/进入根目录,我们需要使用/home/user/命令。
运行
nonhup bash 01.fastp.run.sh三、数据比对
3.1 构建基因组索引
- 基因组下载
本次,我们使用番茄基因组,下载番茄最新基因组,并构建索引文件。
网址:
https://solgenomics.net/organism/solanum_lycopersicum/genome
mkdir 02_Geneome_Tomato
## Fa
wget https://solgenomics.net/ftp/tomato_genome/assembly/build_4.00/S_lycopersicum_chromosomes.4.00.fa
# gff
wget https://solgenomics.net/ftp/tomato_genome/annotation/ITAG4.0_release/ITAG4.0_gene_models.gff
## CDNA
wget https://solgenomics.net/ftp/tomato_genome/annotation/ITAG4.0_release/ITAG4.0_cDNA.fasta
## proteins fa
wget https://solgenomics.net/ftp/tomato_genome/annotation/ITAG4.0_release/ITAG4.0_proteins.fasta
## CDS
wget https://solgenomics.net/ftp/tomato_genome/annotation/ITAG4.0_release/ITAG4.0_CDS.fasta- 构建索引
mkdir Tomato_index_bowtie2
##
## 02.bowtie2_index.sh
time singularity exec ~/SingularityLib/bowtie2.sif bowtie2-build --threads 40 \
Tomato_ref_4.0/S_lycopersicum_chromosomes.4.00.fa Tomato_index_bowtie2/Sl --threads 203.2 Bowtie2序列比对
注意:根据前人教程提示以及我们经常跑数据同学知道,
sam文件占用空间是非常大的。可能会达到几十GB,甚至有些会达到100多GB。因此,结合前人的经验和教程,以及提醒,在bowtie2程序后面加上samtools sort,直接换行成bam文件。用<font style="color:rgb(0, 0, 0);">samtools flagstat</font>看看比对结果。
mkdir 03_mapped**03.bowtie2_mapped.sh**
#!/bin/bash
#
Raw="/home/user/ChipSeq_PRJNA1124576/00_Raw_data"
Clean="/home/user/ChipSeq_PRJNA1124576/01_Clean_Chipsep"
index="/home/user/ChipSeq_PRJNA1124576/02_Geneome_ref/Tomato_index_bowtie2/Sl"
mapped="/home/user/ChipSeq_PRJNA1124576/03_mapped"
#
cat ${Raw}/data.list.txt | awk '{print $1}' | while read id;
do
time singularity exec ~/SingularityLib/bowtie2.sif bowtie2 --threads 40 \
-x ${index} -1 ${Clean}/${id}_1.clean.fastq.gz -2 ${Clean}/${id}_2.clean.fastq.gz | samtools sort -O bam -@ 20 -o ${mapped}/${id}.bam
samtools flagstat ${mapped}/${id}.bam > ${mapped}/${id}_flagstat.txt
done注释:
# bowtie2参数:
# -p 设置线程数
# -x bowtie2索引文件路径及前缀
# -1,-2:双端文件
# samtools sort参数
# -O bam 输出bam格式的文件
# -@ 设置线程数
# -o 输出文件的路径,Single End:
bowtie2 -p 10 -x 02_Geneome_index/Bowtie2-index/Tomato-bowtie-index -U input.fq -S **.sam 2> **.bowtie2.logPaired End:
bowtie2 -p 10 -x 02_Geneome_index/Bowtie2-index/Tomato-bowtie-index -1 **_1.fq.gz -2 **_2.fq.gz -S **.sam 2> **.bowtie2.log可以使用管道符|进行sort排序
bowtie2 -p 10 -x 02_Geneome_index/Bowtie2-index/Tomato-bowtie-index -U input.fq | samtools sort -O bam -@ 10 -o - > output.bam详情可以参考“BioinfoR生信笔记”中转录组零基础教程。
3.3 peak calling
使用<font style="color:rgb(0, 0, 0);">samtools markdup</font>进行PCR去重,再用<font style="color:rgb(0, 0, 0);">samtools flagstat</font>检查
**<font style="color:rgb(0, 0, 0);">04.samtools_markdup.sh</font>**
#!/bin/bash
#使用samtools markdup进行PCR去重,再用samtools flagstat检查
Raw="/home/user/ChipSeq_PRJNA1124576/00_Raw_data"
mapped="/home/user/ChipSeq_PRJNA1124576/03_mapped"
rmdup="/home/user/ChipSeq_PRJNA1124576/03_mapped/0301_rmdup"
##
cat ${Raw}/data.list.txt | awk '{print $1}' | while read id;
do
samtools markdup-@ 20 -r ${mapped}/${id}.bam ${rmdup}/${id}_rmdup.bam
samtools flagstat -@ 20 ${rmdup}/${id}_rmdup.bam > ${rmdup}/${id}_rmdup_flagstat.txt
done以上代码运行不了,可以尝试一下代码代替。
#!/bin/bash
#使用samtools markdup进行PCR去重,再用samtools flagstat检查
Raw="/home/user/ChipSeq_PRJNA1124576/00_Raw_data"
mapped="/home/user/ChipSeq_PRJNA1124576/03_mapped"
rmdup="/home/user/ChipSeq_PRJNA1124576/03_mapped/0301_rmdup"
##
cat ${Raw}/data.list.txt | awk '{print $1}' | while read id;
do
samtools sort -@ 40 -n ${mapped}/${id}.bam | samtools fixmate -@ 40 -m - - \
| samtools sort -@ 40 - | samtools markdup -@ 40 -r - ${rmdup}/${id}_rmdup.bam
samtools flagstat -@ 20 ${rmdup}/${id}_rmdup.bam > ${rmdup}/${id}_rmdup_flagstat.txt
done以上部分全部代码:
#!/bin/bash
#
Raw="/home/kanghuadu/ChipSeq_PRJNA1124576/00_Raw_data"
Clean="/home/kanghuadu/ChipSeq_PRJNA1124576/01_Clean_Chipsep"
Html="/home/user/ChipSeq_PRJNA1124576/01_Clean_Chipsep/html"
index="/home/kanghuadu/ChipSeq_PRJNA1124576/02_Geneome_ref/Tomato_index_bowtie2/Sl"
mapped="/home/kanghuadu/ChipSeq_PRJNA1124576/03_mapped"
rmdup="/home/kanghuadu/ChipSeq_PRJNA1124576/03_mapped/0301_rmdup"
##
cat ${Raw}/data.list.txt | awk '{print $1}' | while read id;
do
## 过滤数据
time singularity exec ~/SingularityLib/fastp.sif fastp -w 16 \
-i ${Raw}/${id}_1.fastq.gz -o ${Clean}/${id}_1.clean.fastq.gz \
-I {Raw}/${id}_2.fastq.gz -O ${Clean}/${id}_2.clean.fastq.gz \
-j ${Html}/${id}.json \
-h ${Html}/${id}.html
##'mapped
time singularity exec ~/SingularityLib/bowtie2.sif bowtie2 -p 30 -x ${index} -1 ${Clean}/${id}_1.clean.fastq.gz -2 ${Clean}/${id}_2.clean.fastq.gz | samtools sort -O bam -@ 20 -o ${mapped}/${id}.bam
samtools flagstat ${mapped}/${id}.bam > ${mapped}/${id}_flagstat.txt
## peak calling
samtools sort -@ 40 -n ${mapped}/${id}.bam | samtools fixmate -@ 40 -m - - \
| samtools sort -@ 40 - | samtools markdup -@ 40 -r - ${rmdup}/${id}_rmdup.bam
samtools flagstat -@ 20 ${rmdup}/${id}_rmdup.bam > ${rmdup}/${id}_rmdup_flagstat.txt
done根据教程,前面的部分与RNA-seq基本一致,但是后续的分析就是与其差异。
查找DNA结合位点

mkdir 04_Rename根据你数据信息表整理出于样本名对应的表格。
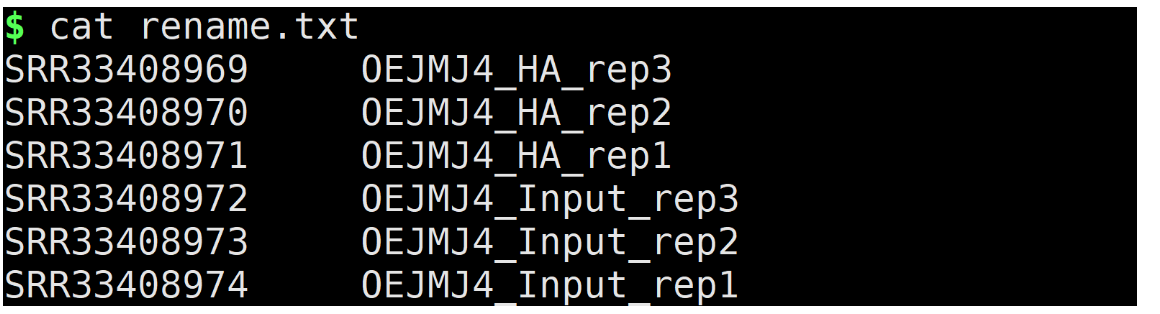
我们基于此信息,整理出一个。
根据生信技能树的推荐,先在本地execl中整理,再粘贴过来。
将整理的文档放如04_Rename文件夹中。
rename.txt
SRR33408969 OEJMJ4_HA_rep3
SRR33408970 OEJMJ4_HA_rep2
SRR33408971 OEJMJ4_HA_rep1
SRR33408972 OEJMJ4_Input_rep3
SRR33408973 OEJMJ4_Input_rep2
SRR33408974 OEJMJ4_Input_rep1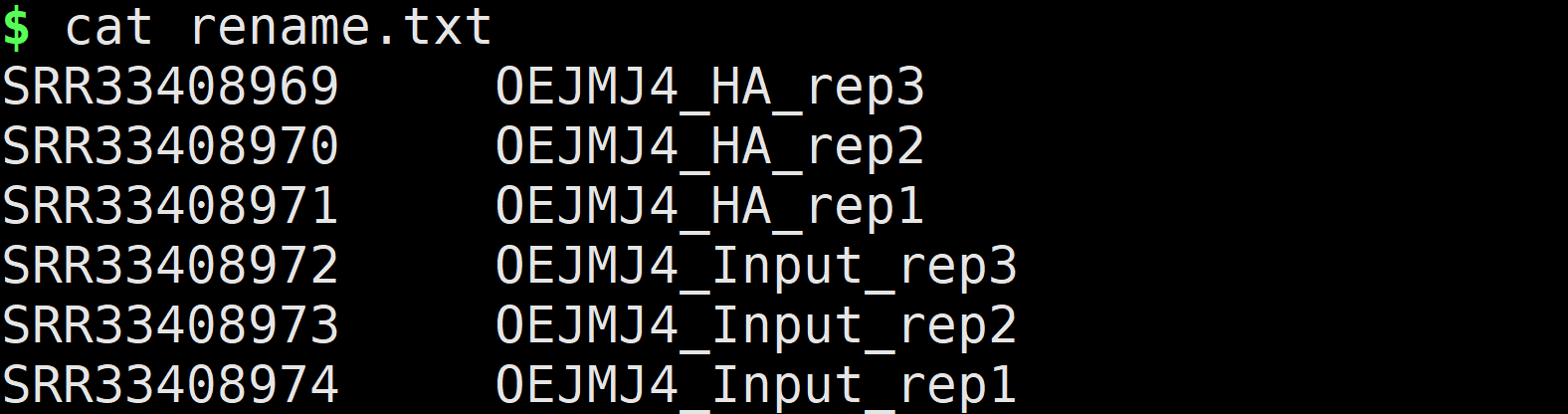
利用上面这个表格,修改样本文件名称:
<font style="color:rgb(0, 0, 0);">05.rename.sh</font>
#!/bin/bash
rmdup="/home/user/ChipSeq_PRJNA1124576/03_mapped/0301_rmdup"
rename_list="/home/user/ChipSeq_PRJNA1124576/04_Rename"
##
cd ${rename_list}
#
for i in $(seq 1 6)
do
id=$(cat rename.txt | awk "{print \$1}" | head -n $i | tail -1)
filename=$(cat rename.txt | awk "{print \$2}" | head -n $i | tail -1)
mv ${rmdup}/${id}_rmdup.bam ${rename_list}/${filename}.bam
samtools sort -@ 20 ${rename_list}/${filename}.bam -o ${rename_list}/${filename}_sort.bam
done合并生物学重复样本
将rename.tsv第二列的_rep后缀去掉,整理一个列表id_norep.txt
OEJMJ4_HA
OEJMJ4_Input用<font style="color:rgb(0, 0, 0);">bedtools intersect</font>将生物学重复样本取交集合并
**<font style="color:rgb(0, 0, 0);">06_bedtools_intersect.sh</font>**
#!/bin/bash
# 用bedtools intersect将生物学重复样本取交集合并
rename_list="/home/user/ChipSeq_PRJNA1124576/04_Rename"
mkdir ${rename_lis}/intersect
##
cat ${rename_list}/id_norep.txt | while read id;
do
time singularity exec ~/SingularityLib/bedtools.sif bedtools intersect \
-a ${rename_list}/${id}_rep1_sort.bam -b ${rename_list}/${id}_rep2_sort.bam ${rename_list}/${id}_rep3_sort.bam > ${rename_list}/intersect/${id}_intersect.bam
samtools index -@ 20 ${rename_list}/intersect/${id}_intersect.bam
donemacs2 callpeak执行peak calling
macs2 peakcall和bamCoverage之间没有先后顺序的要求,这里就先执行macs2 peakcall。
**07.macs2_callpeak.sh**
#!/bin/bash
#macs2 callpeak执行peak calling
rename_list="/home/kanghuadu/ChipSeq_PRJNA1124576/04_Rename"
cd ${rename_list}
mkdir macs2
##
for i in $(seq 1 6)
do
id_HA=$(cat ${rename_list}/id_norep.txt | grep "HA" | head -n ${i} | taill -1)
id_Input=$(cat ${rename_list}/id_norep.txt | grep "Input" | head -n ${i} | tail -1)
time singularity exec ~/SingularityLib/MACS2.sif macs2 callpeak -t ${rename_list}/intersect/${id_HA}_intersect.bam --control ${rename_list}/macs2/OEJMJ4_HA.bam \
-g 7.59e8 --outdir ${rename_list}/macs2/ -n ${id_HA}_intersect -q 0.01
time singularity exec ~/SingularityLib/MACS2.sif macs2 callpeak ${rename_list}/intersect/${id_Input}_intersect.bam --control ${rename_list}/macs2/OEJMJ4_Input.bam \
-g 7.59e8 --outdir ${rename_list}/macs2/ -n ${id_HA}_intersect -q 0.01
done若是小样本直接手动输入即可:
time singularity exec ~/SingularityLib/MACS2.sif macs2 callpeak -t ./intersect/OEJMJ4_HA_intersect.bam \
-g 782475302 --outdir ./macs2/ -n OEJMJ4_HA_intersect -q 0.01# macs2 callpeak参数:
# -t 输入文件
# --control 对照组文件。在本数据集中,共有两个对照组文件——VCaP_EtOH_Control.bam和VCaP_R1881_Control.bam,它们分别是不同处理的对照组。
# -g 基因组大小。输入具体的数字(如1.0e+9或1000000000)。对于人、小鼠、线虫或果蝇,可以分别用hs、mm、ce或dm。
# --outdir 输出文件的路径
# -n 文件名前缀
# -q Q值完成后,得到下面一堆文件。每个样本有4个文件,包括macs2 callpeak返回4个结果文件:<font style="color:rgb(0, 0, 0);">id_peaks.narrowPeak</font>,<font style="color:rgb(0, 0, 0);">id_peaks.xls</font>,<font style="color:rgb(0, 0, 0);">id_summits.bed</font>和<font style="color:rgb(0, 0, 0);">id_model.r</font>。
Rscript OEJMJ4_HA_intersect_model.r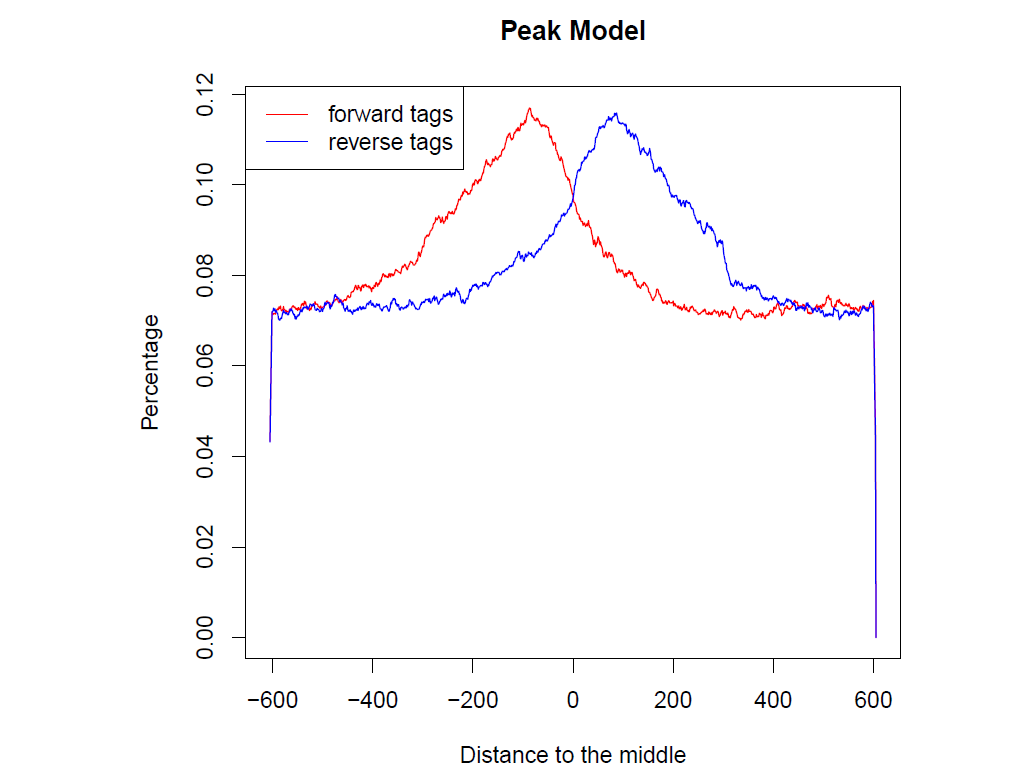

引用以前推文中的解释
bamCoverage将bam文件转化为反映基因组区域reads覆盖度结果的bw文件
bw文件同样可用于后续在IGV中可视化,而且bw格式的文件更小,便于存储。

mkdir 05_bamCoverage08.bam_To_bw.sh
#!/bin/bash
bw="/home/kanghuadu/ChipSeq_PRJNA1124576/05_bamCoverage"
rename_list="/home/user/ChipSeq_PRJNA1124576/04_Rename"
#
for i in $(seq 1 6)
do
id_HA=$(cat ${rename_list}/id_norep.txt | grep "HA" | head -n ${i} | taill -1)
id_Input=$(cat ${rename_list}/id_norep.txt | grep "Input" | head -n ${i} | tail -1)
bamCoverage --binSize 10 -p 40 --normalizeUsing RPKM -b ${rename_list}/intersect/${id_HA}_intersect.bam -o ${bw}/${id_HA}.bw
bamCoverage --binSize 10 -p 40 --normalizeUsing RPKM -b ${rename_list}/intersect/${id_Input}_intersect.bam -o ${bw}/${id_Input}.bw
done| 参数 | 说明 |
|---|---|
-b | 输入 BAM 文件(必须) |
-o | 输出文件名,通常用 .bw 或 .bigwig |
--outFileFormat | 输出格式,bigwig 或 bedgraph,默认根据后缀自动判断 |
-p / --numberOfProcessors | 使用的线程数,提高处理速度 |
--binSize | 输出信号每个 bin 的大小(单位 bp),默认 50,越小越精细但文件大 |
--normalizeUsing | 归一化方法,可选:RPKM、CPM、BPM、RPGC(Reads Per Genomic Content) |
--scaleFactor | 手动归一化因子(可与 --normalizeUsing None 配合) |
--ignoreDuplicates | 忽略 BAM 中标记的重复 reads |
--minMappingQuality | 最小比对质量(MAPQ)过滤,例如 30 |
--extendReads | 将 reads 延长到指定长度(常用于单端 ChIP-seq 数据) |
--effectiveGenomeSize | 用于 RPKM/RPGC 归一化,需要指定物种有效基因组大小 |
--smoothLength | 平滑窗口长度,可用于降低噪声 |
--centerReads | 将 reads 居中(通常用于单端 ChIP-seq) |
--filterRNAstrand | 对 RNA-seq 数据,按链选择正/负链 |
--ignoreForNormalization | 排除特定染色体用于归一化(如 chrM、chrY) |
常用示例
- 生成 BigWig,默认 binSize=50,归一化为 CPM,单端数据
bamCoverage -b sample.bam -o sample.bw -p 8 --normalizeUsing CPM- 生成 BigWig,单端 ChIP-seq,延长 reads 到 200 bp,去重复
bamCoverage -b sample.bam -o sample.bw -p 8 \
--binSize 25 --extendReads 200 --ignoreDuplicates \
--normalizeUsing RPGC --effectiveGenomeSize 759000000- 生成 BedGraph 文件
bamCoverage -b sample.bam -o sample.bedgraph --outFileFormat bedgraph**注意事项**
- 有效基因组大小 (
**--effectiveGenomeSize**)- 对非人类/小鼠物种必须手动指定,例如番茄 ~7.59e8。
- 线程数 (
**-p**)- 对大 BAM 文件必加,否则会很慢。
- 单端 vs 双端
- 双端 BAM 默认不需要
--extendReads。
- 双端 BAM 默认不需要
- 归一化方法
- RPKM/RPGC 会考虑基因组大小;CPM/CPM-like 只做 counts per million。
我们使用(https://mp.weixin.qq.com/s/CzIO7lepe9umosBE2BCrsA)的流程分析。
-I ${Raw}/${id}_2.fastq.gz -O ${wd}/01_Clean_Chipsep/${id}_2.clean.fastq.gz \
-j ${wd}/01_Clean_Chipsep/html/${id}.json \
-h ${wd}/01_Clean_Chipsep/html/${id}.html
time singularity exec ~/SingularityLib/bowtie2.sif bowtie2 -p 40 \
-x ${index} \
-1 ${wd}/01_Clean_Chipsep/${id}_1.clean.fastq.gz \
-2 ${wd}/01_Clean_Chipsep/${id}_2.clean.fastq.gz \
-S ${wd}/03_mapped/${id}.sam \
1>${wd}/logs/${id}_align.log 2>&1
## peak calling
samtools sort -@ 40 -o ${wd}/03_mapped/0301_sort/${id}.sort.bam ${wd}/03_mapped/${id}.sam 1>${wd}/logs/${id}_sort_bam.log 2>&1
samtools index -@ 40 ${wd}/03_mapped/0301_sort/${id}.sort.bam
## 过滤
alignmentSieve --numberOfProcessors 40 --bam ${wd}/03_mapped/0301_sort/${id}.sort.bam \
--outFile ${wd}/03_mapped/0302_clean_bam/${id}.final.bam \
--filterMetrics ${wd}/03_mapped/0302_clean_bam/${id}.sieve_alignment.log \
--ignoreDuplicates \
--minMappingQuality 25 \
--samFlagExclude 260
samtools index -@ 40 ${wd}/03_mapped/0302_clean_bam/${id}.final.bam
##bam to bw
bamCoverage -p 40 \
--bam ${wd}/03_mapped/0302_clean_bam/${id}.final.bam \
-o ${wd}/04_bw/${id}.bw \
--normalizeUsing RPKM \
--binSize 50 \
--effectiveGenomeSize 782475302 \
1>${wd}/04_bw/${id}_bam_to_bigwig.log 2>&1
done此外,我们也可以参考教程中的循环流程。
#!/bin/bash
# 样本列表,可以根据需要添加更多样本ID
samples=(
"SRR7444033"
"SRR7444034"
"SRR7444035"
# 在这里添加更多样本ID
)
# 循环处理每个样本
for sample in"${samples[@]}"; do
fastp -i "raw_data/${sample}_R1.fastq.gz" \
-o "clean_data/${sample}_R1.fastq.gz" \
--html "clean_data/${sample}_fastp.html" \
--json "clean_data/${sample}_fastp.json" \
--thread 4 \
--qualified_quality_phred 20 \
--length_required 50 \
1>"logs/${sample}.log" 2>&1
done计算基因组大小python脚本
#coding=utf-8
import sys
aList=[]
fa_file = sys.argv[1]
with open(fa_file,'r') as f:
for line in f:
line = line.strip()
line = line.upper()
if not line.startswith(">"):
baseA = line.count("A")
baseT = line.count("T")
baseC = line.count("C")
baseG = line.count("G")
aList.extend([baseA, baseT, baseC, baseG])
# print(aList)
print("effective_genome_size =", sum(aList))运行
python genomeSize.py genome.fa绘制peak分布热图
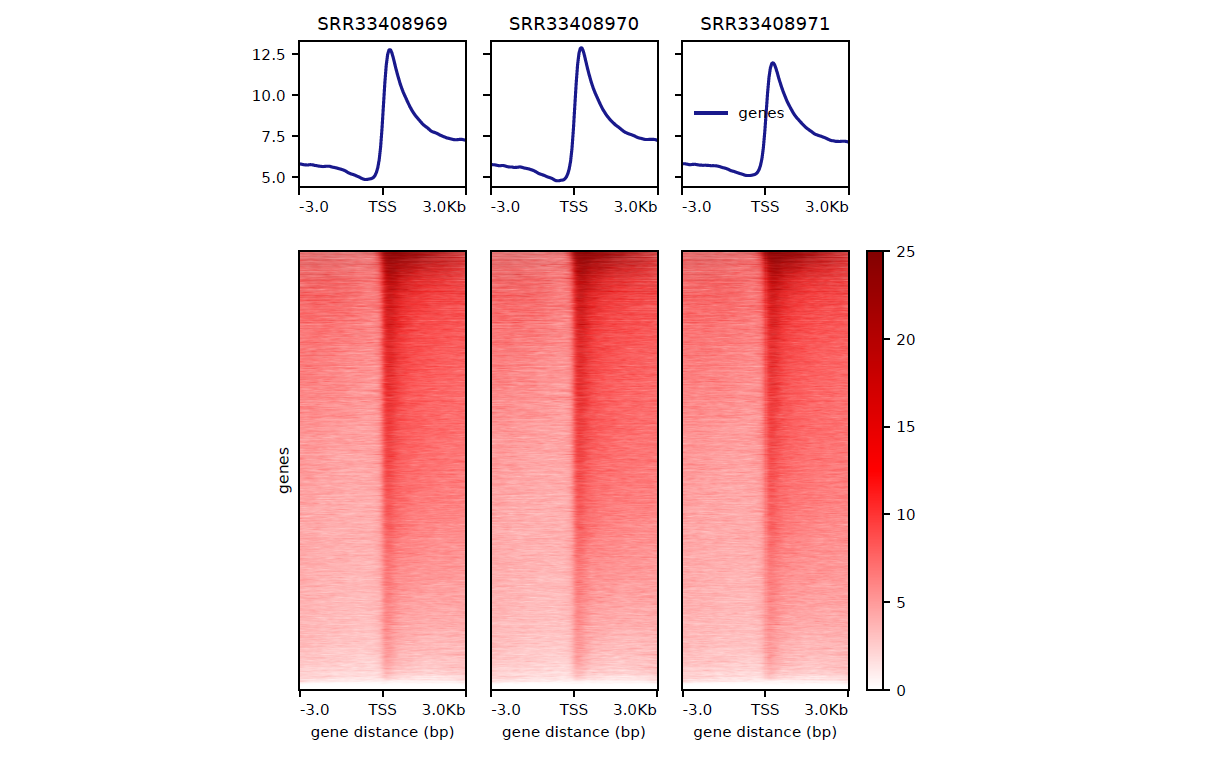
**<font style="color:rgb(5, 145, 255);">reference-point</font>**模式:以特定坐标(TSS)为参考,提取上下游固定长度的信号。
gtf="/home/kanghuadu/ChipSeq_PRJNA1124576/02_Geneome_ref/Tomato_ref_4.0/ITAG4.0_gene_models.gtf"
## reference-point 模式:以特定坐标(TSS)为参考,提取上下游固定长度的信号。
time computeMatrix reference-point \
-S ${wd}/04_bw/SRR33408969.bw ${wd}/04_bw/SRR33408970.bw ${wd}/04_bw/SRR33408971.bw \
--samplesLabel SRR33408969 SRR33408970 SRR33408971 --missingDataAsZero \
-R ${gtf} \
--referencePoint TSS \
-b 3000 -a 3000 \
--binSize 50 \
-o ${wd}/TSS/HA_tss_matrix.gz \
--numberOfProcessors 40**<font style="color:rgb(5, 145, 255);">reference-point</font>**模式:以特定坐标(TES)为参考,提取上下游固定长度的信号。
gtf="/home/kanghuadu/ChipSeq_PRJNA1124576/02_Geneome_ref/Tomato_ref_4.0/ITAG4.0_gene_models.gtf"
## TES模式
#reference-point 模式:以特定坐标(TES)为参考,提取上下游固定长度的信号
time computeMatrix reference-point \
-S ${wd}/04_bw/SRR33408969.bw ${wd}/04_bw/SRR33408970.bw ${wd}/04_bw/SRR33408971.bw \
--samplesLabel SRR33408969 SRR33408970 SRR33408971 --missingDataAsZero \
-R ${gtf} \
--referencePoint TES \
-b 3000 -a 3000 \
--binSize 50 \
-o ${wd}/TES/HA_tes_matrix.gz \
--numberOfProcessors 40**<font style="color:rgb(5, 145, 255);">scale-regions</font>**模式:将所有区域缩放至相同长度,适合比较不同长度区域的信号分布。
bed="/home/kanghuadu/ChipSeq_PRJNA1124576/02_Geneome_ref/Tomato_ref_4.0/ITAG4.0_gene_models.bed"
time computeMatrix scale-regions \
-S ${wd}/04_bw/SRR33408969.bw ${wd}/04_bw/SRR33408970.bw ${wd}/04_bw/SRR33408971.bw \
--samplesLabel SRR33408969 SRR33408970 SRR33408971 --missingDataAsZero \
-R ${bed} \
--regionBodyLength 4000 \
-b 2000 -a 2000 \
--binSize 50 \
-o ${wd}/Scale_regions/HA_tss_tes_matrix.gz \
--numberOfProcessors 40bed文件使用gtf文件制作即可。
SL4.0ch00 93749 94430 Solyc00g500001.1 . +
SL4.0ch00 305441 306257 Solyc00g500002.1 . -
SL4.0ch00 311495 382066 Solyc00g500003.1 . -
SL4.0ch00 417591 418482 Solyc00g500004.1 . +
SL4.0ch00 478388 478640 Solyc00g500005.1 . +
SL4.0ch00 481287 483182 Solyc00g500006.1 . -
SL4.0ch00 497940 498614 Solyc00g500007.1 . +
SL4.0ch00 531007 531682 Solyc00g500008.1 . +
SL4.0ch00 531760 537711 Solyc00g500009.1 . +
SL4.0ch00 544693 545489 Solyc00g500010.1 . +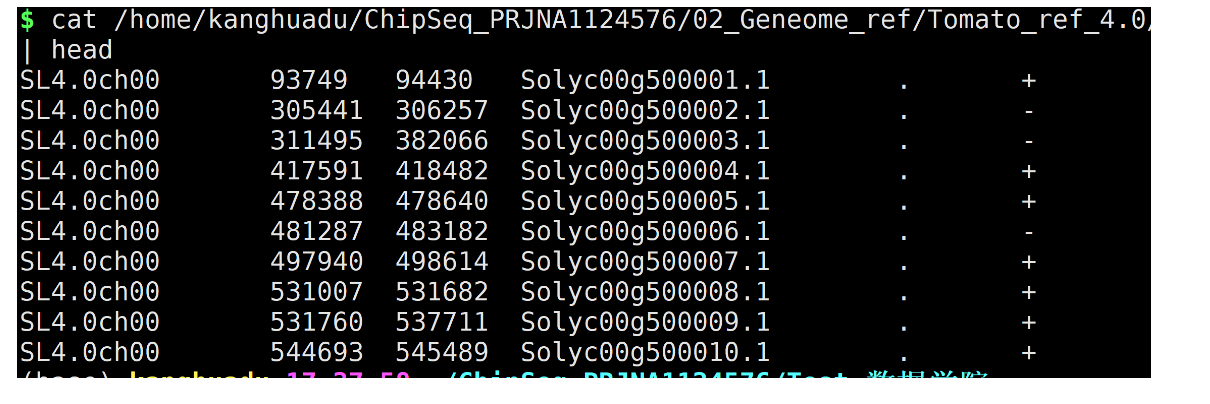
绘制热图
## 使用 plotHeatmap 将矩阵数据可视化为热图,支持聚类、颜色调整和注释。
## TSS
plotHeatmap -m ${wd}/TSS/HA_tss_matrix.gz -o ${wd}/TSS/HA_tss.pdf --colorList 'white,red,#830101' --heatmapWidth 4 --heatmapHeight 12
## TES
plotHeatmap -m ${wd}/TES/HA_tes_matrix.gz -o ${wd}/TES/HA_tes.pdf --colorList 'white,red,#830101' --heatmapWidth 4 --heatmapHeight 12
## scale-regions
plotHeatmap -m ${wd}/Scale_regions/HA_tss_tes_matrix.gz -o ${wd}/Scale_regions/HA_tss_tes.pdf --colorList 'white,red,#830101' --heatmapWidth 4 --heatmapHeight 12**plotHeatmap图形的宽与高参数**
--heatmapWidth 宽
--heatmapHeight 高
其它参数优化
- 聚类与排序:
<font style="color:rgb(5, 5, 5);">--kmeans 4</font>:将区域分为 4 个聚类。<font style="color:rgb(5, 5, 5);">--clusteringMethod ward.D2</font>:指定层次聚类方法。
- 颜色与范围:
<font style="color:rgb(5, 5, 5);">--zMin 0 --zMax 10</font>:限制颜色显示范围,避免极端值影响视觉效果。
- 布局与注释:
<font style="color:rgb(5, 5, 5);">--heatmapHeight 10</font>:调整热图高度(单位:cm)。<font style="color:rgb(5, 5, 5);">--regionsLabel "Promoters"</font>:添加区域标签。<font style="color:rgb(5, 5, 5);">--samplesLabel "Ctrl" "Treat"</font>:指定样本名称。
联合 plotProfile 可视化
## plotProfile 可视化
## TSS
plotProfile --matrixFile ${wd}/TSS/HA_tss_matrix.gz \
--outFileName ${wd}/TSS/HA_tss.profile.pdf \
--perGroup # 按分组绘制曲线
## TES
plotProfile --matrixFile ${wd}/TES/HA_tes_matrix.gz \
--outFileName ${wd}/TES/HA_tes.profile.pdf \
--perGroup
## scale-regions
plotProfile --matrixFile ${wd}/Scale_regions/HA_tss_tes_matrix.gz \
--outFileName ${wd}/Scale_regions/HA_tss_tes.profile.pdf \
--perGroup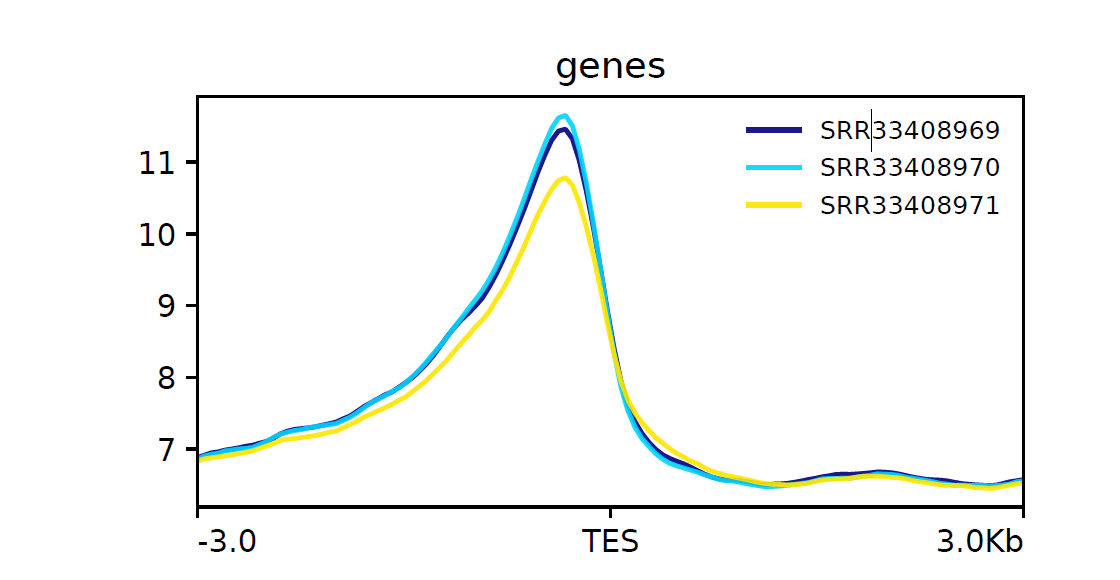
全部代码如下:
gtf="/home/kanghuadu/ChipSeq_PRJNA1124576/02_Geneome_ref/Tomato_ref_4.0/ITAG4.0_gene_models.gtf"
## reference-point 模式:以特定坐标(TSS)为参考,提取上下游固定长度的信号。
time computeMatrix reference-point \
-S ${wd}/04_bw/SRR33408969.bw ${wd}/04_bw/SRR33408970.bw ${wd}/04_bw/SRR33408971.bw \
--samplesLabel SRR33408969 SRR33408970 SRR33408971 --missingDataAsZero \
-R ${gtf} \
--referencePoint TSS \
-b 3000 -a 3000 \
--binSize 50 \
-o ${wd}/TSS/HA_tss_matrix.gz \
--numberOfProcessors 40
## TES模式
#reference-point 模式:以特定坐标(TES)为参考,提取上下游固定长度的信号
time computeMatrix reference-point \
-S ${wd}/04_bw/SRR33408969.bw ${wd}/04_bw/SRR33408970.bw ${wd}/04_bw/SRR33408971.bw \
--samplesLabel SRR33408969 SRR33408970 SRR33408971 --missingDataAsZero \
-R ${gtf} \
--referencePoint TES \
-b 3000 -a 3000 \
--binSize 50 \
-o ${wd}/TES/HA_tes_matrix.gz \
--numberOfProcessors 40
# scale-regions 模式:将所有区域缩放至相同长度,适合比较不同长度区域的信号分布
bed="/home/kanghuadu/ChipSeq_PRJNA1124576/02_Geneome_ref/Tomato_ref_4.0/ITAG4.0_gene_models.bed"
time computeMatrix scale-regions \
-S ${wd}/04_bw/SRR33408969.bw ${wd}/04_bw/SRR33408970.bw ${wd}/04_bw/SRR33408971.bw \
--samplesLabel SRR33408969 SRR33408970 SRR33408971 --missingDataAsZero \
-R ${bed} \
--regionBodyLength 4000 \
-b 2000 -a 2000 \
--binSize 50 \
-o ${wd}/Scale_regions/HA_tss_tes_matrix.gz \
--numberOfProcessors 40
## 使用 plotHeatmap 将矩阵数据可视化为热图,支持聚类、颜色调整和注释。
## TSS
plotHeatmap -m ${wd}/TSS/HA_tss_matrix.gz -o ${wd}/TSS/HA_tss.pdf --colorList 'white,red,#830101'
## TES
plotHeatmap -m ${wd}/TES/HA_tes_matrix.gz -o ${wd}/TES/HA_tes.pdf --colorList 'white,red,#830101'plotHeatmap图形的宽与高参数
--heatmapWidth 宽
--heatmapHeight 高若我们的教程对你有所帮助,请点赞+收藏+转发,大家的支持是我们更新的动力!!
2024已离你我而去,2025加油!!
往期部分文章
1. 最全WGCNA教程(替换数据即可出全部结果与图形)
推荐大家购买最新的教程,若是已经购买以前WGNCA教程的同学,可以在对应教程留言,即可获得最新的教程。(注:此教程也仅基于自己理解,不仅局限于此,难免有不恰当地方,请结合自己需求,进行改动。)
2. 精美图形绘制教程
3. 转录组分析教程
4. 转录组下游分析
BioinfoR生信筆記 ,注于分享生物信息学相关知识和R语言绘图教程。


 浙公网安备 33010602011771号
浙公网安备 33010602011771号