
2Developing Nonstandard Parameters: 2.4 Metal Ion Modeling Tutorial
- Metal Ion Modeling Tutorial
包含了Bonded Model,Nonbonded Model以及Cationic Dummy Atom Model
其中Bonded Model包含了两种方法,一种是MCPB;另一种是它的python版本MCPB.py,其优化了工作流,并使用较少的步骤进行建模 (Needs AmberTools 15 or Higher)。The current version supports more than 80 metal ions and various AMBER force fields. It was developed by Pengfei Li in the Merz research group in Michigan State University.
这里介绍的是MCPB.py的其中一个例子:Building Bonded Model for A Ligand Binding Metalloprotein with MCPB.py
(另外,关于MCPB.py还有一个例子:Building Bonded Model for A HEME Group with MCPB.py——https://ambermd.org/tutorials/advanced/tutorial20/mcpbpy_heme.php,这是构建HEM力场的例子,其中FE离子bond到卟啉环的4个N原子上和一个CYS残基的S原子上。构建的过程和当前的内容差不多。)
(另外,如果使用该成键模型方法得到的模型效果不好,可以使用局部非键模型:Building An Restrained Nonbonded Model——https://ambermd.org/tutorials/advanced/tutorial20/rnb.htm。构建非键模型(或者说局部非键模型,即某些键使用成键某些建使用非键)的过程也是需要先使用MCPB.py计算整体成键模型,然后在tLeap的时候选定减少一些添加的成键bond,不添加bond的这些键长使用harmonic restraints进行距离限制,其中限制的时候平衡键长和力常数都从上一步MCPB.py计算成键模型时的结果取出。)
使用MCPB.py处理金属体系一共有三种选择(以前用的第三种方案是最不靠谱的,只能施加键长键角限制金属的相对位置):

正文如下:
在这里,我们以锌酶系统为例(PDB ID:1OKL),其金属部位有3个His残基和1个MNS(5-(二甲氨基)-1-萘磺酰胺)配体。
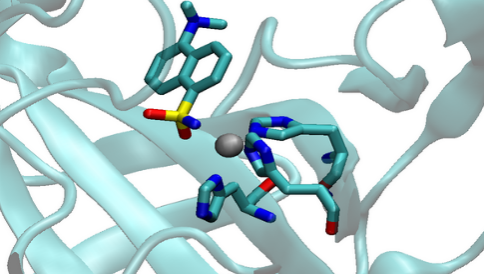
1.准备PDB文件和mol2文件
1)首先准备非标准残基的PDB和mol2文件
①配体
reduce MNS.pdb > MNS_H.pdb
然后删除一个氢原子,即配体中与锌离子配位的N原子上的一个氢原子HN32,得到MNS_fixed_H.pdb
然后执行:antechamber -fi pdb -fo mol2 -i MNS_fixed_H.pdb -o MNS_pre.mol2 -c bcc -pf y -nc -1
检查MNS_pre.mol2文件,发现没有“du”原子类型。因此可以将其改名为MNS.mol2.
然后生成frcmod文件:parmchk2 -i MNS.mol2 -o MNS.frcmod -f mol2 #这里我们使用了parmchk2,因为它比parmchk有更好的性能
②金属离子
使用脚本metalpdb2mol2.py生成mol2文件,如果是AmberTools20,那么可以直接:
metalpdb2mol2.py -i ZN.pdb -o ZN.mol2 -c 2
此处“-c 2”表示离子的电荷为2.0。 得到ZN.mol2.
2)准备包含所有标准残基的PDB文件
我们可以使用the webserver H++ (here we use the default settings)为PDB加上氢原子。H++将在建模过程中删除非标准残基。除了生成带有氢原子的PDB文件外,它还将为系统生成amber的拓扑和坐标文件。然后我们可以使用拓扑和坐标文件生成PDB文件,因此这个PDB文件将使用amber的残基明命名方案(比如组氨酸使用HID,HIE,HIP而不是HIS)。这里执行以下命令:
ambpdb -p 0.15_80_10_pH6.5_1OKL.top -c 0.15_80_10_pH6.5_1OKL.crd > 1OKL_Hpp.pdb
H++在添加氢原子的时候不考虑金属离子。我们发现金属位点有一个HIP残基,这没有物理意义。所以我们这里手动将HIP-92修改为HID-92(通过删除HE2原子并将该残基名称从HIP改为HID),修改后的文件为1OKL_Hpp_fixed.pdb
PS:实际做的时候H++网站会崩溃,我直接使用的amber的默认质子化状态,然后手动修改ZN离子周围的残基质子化状态。
3)最终我们combine成一个整体的PDB文件
为了与PDB文件中的常规顺序一致,我们首先放置标准残基,然后放置金属离子,然后放置配体,然后放置水(如果有)
cat 1OKL_Hpp_fixed.pdb ZN.pdb MNS_fixed_H.pdb > 1OKL_H.pdb
然后使用pdb4amber对PDb文件重新编号:
pdb4amber -i 1OKL_H.pdb -o 1OKL_fixed_H.pdb
现在我们有了PDB文件1OKL_fixed_H.PDB和mol2文件MNS.mol2和ZN.mol2用于建模的下一阶段。
2、生成PDB、Gaussian和指纹建模文件:
我们需要手动创建输入文件。这是输入文件1OKL.in。可以查阅手册了解MCPB使用的输入参数。
然后执行:MCPB.py -i 1OKL.in -s 1
在此步骤中,将生成小型、标准和大型模型的PDB和指纹文件。还将生成小模型和大模型的高斯输入文件。大模型的高斯输入文件将首先对氢原子进行优化,以纠正任何位置不当的氢原子。标准模型指纹文件(1OKL_standard.fingerprint)包含标准模型中原子的原子类型信息(第3列为原始原子类型,最后一列为最终指定的原子类型)。金属离子的原子类型(名称以M开头)和连接原子(名称以Y/Z开头)将自动重命名,以区别于amber力场中的其他原子类型。指纹文件的末尾还包含金属离子和周围原子之间的链接信息,开头字母为“LINK”。用户还可以根据自己的需要手动修改它们(例如,如果QM几何优化后存在任何连键更改)。
3、执行高斯计算
①执行几何优化,然后计算力常数,然后为小模型生成fchk文件
g16 < 1OKL_small_opt.com > 1OKL_small_opt.log #这个结果计算完成后才能进行后面两个计算(后面两个需要读取1OKL_small_opt.chk中的信息)
g16 < 1OKL_small_fc.com > 1OKL_small_fc.log
formchk 1OKL_small_opt.chk 1OKL_small_opt.fchk
②对大模型执行Merz-Kollman RESP电荷计算
g16 < 1OKL_large_mk.com > 1OKL_large_mk.log
#!这第3步是最容易出问题的地方,如不收敛等:可以尝试调整方法,基组,自选多重度等。。。比如我在计算含有一个FE3+和一个带电为-1的GLU的体系的时候,电荷为+2,把自选多重度定为2的话可以正常计算,但是结果不收敛,如果把自旋多重度定为4的话,就可以计算且收敛。 具体应该用哪个自旋多重度,Sob老师解释说可以最低的几个自旋多重度都计算一下,用能量最低的。作者也说了:“We note that defining the total charge and spin state is a complicated problem, one may need to adapt them for different situations.”
4、执行最终建模
①在此,我们使用Seminario方法生成力场参数。其他选项可用:Z矩阵(with step_number 2z)和经验(with step_number 2e)方法。经验方法不需要任何高斯计算来获得力常数(但仍然需要高斯计算来获得RESP电荷),但它仅支持当前版本中的锌离子建模。
MCPB.py -i 1OKL.in -s 2
我们得到了1OKL_mcpbpy.frcmod文件,将在leap建模中使用
②在此,我们使用ChgModB执行RESP电荷拟合,并生成金属位置残基的mol2文件。其他选项也可用:ChgModA、ChgModC和ChgModD(分别为3a、3c和3d)。
MCPB.py -i 1OKL.in -s 3
在这一步中,我们可以得到金属位点的5个mol2文件:HD1.mol2, HD2.mol2, HE1.mol2, ZN1.mol2 and MS1.mol2. 这些文件中的电荷由MK RESP电荷拟合算法重新调整。我们将在leap建模中使用这些mol2文件。
③生成tleap输入文件
MCPB.py -i 1OKL.in -s 4
然后我们可以得到一个新的PDB文件(1OKL_mcpbpy.pdb)(重命名了金属位点残基名)和一个leap输入文件(1OKL_tleap.in)。我们可以根据需要更改这个输入文件。 注意!:这里最好是保持蛋白力场符合前面使用的AmberTools版本(即使用AmberTools自带的蛋白力场)!不然可能会出现某些原子类型缺失(实测发现/opt/amber16/dat/leap/lib/amino12.lib中HID的CA的原子类型是CX,而.software/anaconda3/envs/AmberTools20/dat/leap/lib/amino19.lib中HID的CA的原子类型是XC)!
④生成拓扑和坐标文件
tleap -s -f 1OKL_tleap.in > 1OKL_tleap.out
得到了( 1OKL_solv.prmtop and 1OKL_solv.inpcrd),可以用于下一步的MD。
5、检查模型!
1)导入VMD查看键连方式
2)执行cpptraj -p 1OKL_solv.prmtop看看有没有报错
本文来自博客园,作者:计算之道,转载请注明原文链接:https://www.cnblogs.com/jszd/p/15725985.html

 浙公网安备 33010602011771号
浙公网安备 33010602011771号