扩增子分析解读1质控 实验设计 双端序列合并
本文采用目前最主流的扩增子测序数据类型HiSeq2500 PE250类型数据为例,结合目前主流方法QIIME+USearch定制的分析流程。本课程中所需的测序数据、实验设计和课程分析生成的中间文件,均可以直去百度云下载。链接:http://pan.baidu.com/s/1hs1PXcw 密码:y33d
本课程代码的运行,至少需要Linux平台+安装QIIME 1
分析前准备
# 建立工作目录并进入,-p参数为如果文件夹存在不报错
mkdir -p example_PE250 cd example_PE250
# 建临时文件和结果子目录
mkdir -p temp result
1. 测序数据文件
16S扩增子测序数据主要来自HiSeq2500产出的双端各250 bp (PE250)数据,因为读长长且价格便宜(性价比高)。HiSeqX PE150和MiSeq PE300也比较常见,但PE150过短分辨率低,而PE300价格高且末端序列质量过低。此外454在之前研究较多但设备已经停产,PacBio读长长可直接测序16S全长1.5kb代表未来的趋势。
测序公司通常会返回raw data和clean data两种数据,raw data为测序获得的原始数据,而clean data则为去除含有接头序列及测序不确定N比例较高的结果,通常直接采用clean data进行质量评估及后续分析。
质量评估常用fastqc,一般测序结果文件会附带评估报告,质量太差会重测,此步非用户必须
准备两个数据文件PE250_1.fq.gz和PE250_2.fq.gz至工作目录,一共600M,包括2,500,000条fastq格式的双端250bp数据。(提示:可以在Windows上下载,使用filezilla等工具上传服务器)
安装fastqc,己安装请跳过,未安装详见http://www.cnblogs.com/freescience/p/7277556.html
如果系统中己安装过fastqc可直接运行fastqc -t 2 *.fq.gz即可。-t为设置线程数,建议与数据文件数量相同最佳,可以提高评估速度,*.fq.gz为输入文件,可以用*通配符指定多个文件。
运行结果每个数据会生成两个文件,如下
PE250_1_fastqc.html # 网页评估报告PE250_1_fastqc.zip # 网页报告相关文本和图片压缩包
数据质量如下:上为左端1-250质量;下为右端1-250质量分布箱线图
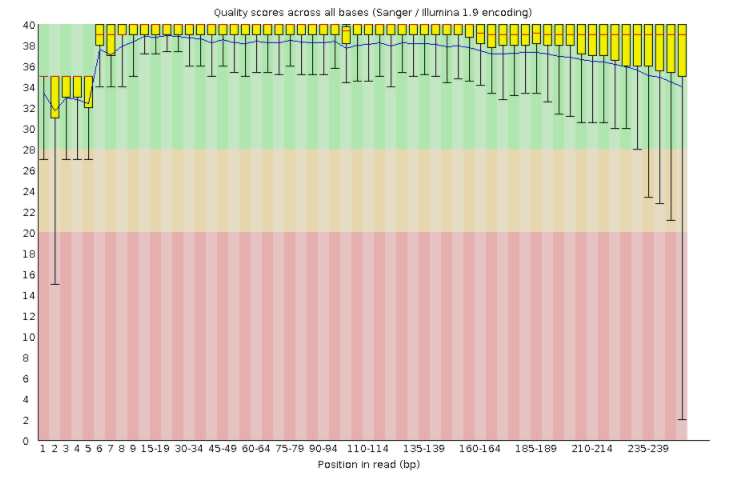

可以看到左端的质量比较高(图中绿、黄、红区域分别代表质量优、良、差);右端序列末端质量较次,且箱体也进入红色差区,但中位数红线位于绿色高质量区。这样的结果已经算是中等偏上的了,在PE250测序中,右端的尾部质量都下降很严重,但只要左端的末端较好即可,双端序列合并可进行校正,一般都可以放心使用。
2. 实验设计文件
在QIIME中,把实验设计文件叫mappingfile,大家下载mappingfile.txt文件;自己的实验一定要按照示例的格式模仿填写,如错误后续无法运行。QIIME自带了个工具,可以检验文件书写是否正确。
# 先激活工作环境 source activate qiime1 # 关闭工作环境:不用时关闭,不然你其它程序可能会出错 source deactivate # 验证实验设计是否有错误 validate_mapping_file.py -m mappingfile.txt
运行结果会输出三个文件
mappingfile_corrected.txt # 自动修正的实验设计,小错误会自动修改,但末必符合你的要求,不建议直接使用mappingfile.html # 结果的错误报告,可下载查看网页,会高亮显示错误的位置mappingfile.log # 运行结果报告
运行结果无误会显示 “No errors or warnings were found in mapping file.”。有错误建议查看生成的网页报告,高亮有错误的地方,自行修改后重新检测,直到无误。更多说明建议阅读帮助http://qiime.org/scripts/validate_mapping_file.html
3. 双端序列合并
我们首先的任务是把双端序列合并,根据两端序列末端的互补配对,可以合变为我们扩增区域的序列,同时还可以对重叠区的质量进行校正,保留最高测序质量的碱基结果。使用join_paired_ends.py脚本,合并两个文件为单个。f/r参数为输入左和右端序列,支持压缩格式*.gz;m是选择方法,默认为fastq-join就可以了,也可以选择SeqPrep,更好但更慢;o为输出文件目录。更多说明建议阅读帮助 http://qiime.org/scripts/join_paired_ends.html
# 双端序列合并 join_paired_ends.py -f PE250_1.fq.gz -r PE250_2.fq.gz -m fastq-join -o temp/PE250_join
序列合并完,我们会在设置的输出目录temp/PE250_join看到3个文件,如下:
fastqjoin.join.fastq # 合并成功的序列fastqjoin.un1.fastq # 左端未合并成功的序列fastqjoin.un2.fastq # 右端未合并成功的序列
我们下游分析通常只对fastqjoin.join.fastq进行操作

 浙公网安备 33010602011771号
浙公网安备 33010602011771号