文献阅读 | Rapid genotype imputation from sequence with reference panels
Davies, R. W., Kucka, M., Su, D., Shi, S., Flanagan, M., Cunniff, C. M., Chan, Y. F., & Myers, S. (2021). Rapid genotype imputation from sequence with reference panels. Nature Genetics. https://doi.org/10.1038/s41588-021-00877-0
廉价的基因分型方法对现代基因组学至关重要。作者开发了QUILT工具,对低覆盖率的全基因组测序数据执行二倍体基因型填充。通过利用吉布斯采样,QUILT首先将读段划分为母本集和父本集,再基于大型参考群体进行快速填充。作者证明,这种划分方法在很多兆碱基对尺度上是准确的,能够实现接近理论极限的高精度填充,并且优于现有方法。
QUILT支持多种测序数据,包括Oxford Nanopore Technologies测得的长读段,和一种称为haplotagging的新的低成本barcoded Illumina sequencing方法。后者在低覆盖率下填充准确率更高。
相较于DNA genotyping microarrays,QUILT能以更低的价格提供更高的准确率。特别是对于之前在现代基因组分析中underserved的差异群体,在罕见的snp中精确度几乎翻了一番。最后,QUILT 可以准确地推断人类白细胞抗原类型,并且是第一个从低覆盖率序列数据中推断的方法。
模型
通过利用Li and Stephens model[1],QUILT首先将每个样本待填充的单倍型建模为一个ref haplotypes的马赛克形式。令\(K\)代表ref haplotypes的数量。由于样本是二倍体(diploid),因此对于该模型的朴素实现(naïve implementation)将具有与\(K^2\)成正比的计算复杂度,而这在\(K\)很大时计算上令人望而却步。然而从根本上讲,每个测序读段都来自母本或父本染色体,因此如果我们可以将它们分成两个这样的集合,那么以填充的计算量将会变成\(K\)的线性关系。
在之前的STITCH模型中,通过为每个读段分配来自每个遗传背景的近似概率,作者引入了一种具有线性计算复杂度的“pseudo-haploid”方法。在本研究中,作者通过有效地重用存储的前向后向概率实现了Gibbs采样器,进而改进了该方法,允许使用隐马尔可夫模型(HMM)的单向前-向后传递将所有读段标签重新采样到两个亲本染色体中。
由于使用全部参考群体时Gibbs采样速度较慢,作者采用了一种迭代方法进行采样:在使用参考群体的子集上利用的Gibbs采样器更新读段标签,并利用读段标签和全部参考群体进行单倍体填充以更新参考群体子集,并在最终一轮迭代中执行真正填充(图1)。通过使用群体的一小部分,Gibbs 采样速度很快;同时通过利用最佳匹配的单倍型进行填充,该方法也是准确的。最终的填充结果取多个吉布斯过程的平均值,整个算法具有\(K\)的线性计算复杂度。
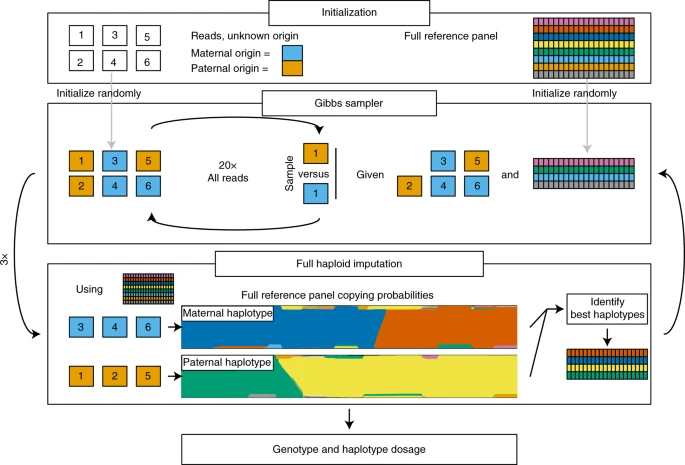
Fig. 1: Schematic of the QUILT model.
The model for one Gibbs sampling is shown. The model is initialized for a vector of read labels and a subset of reference haplotypes. The QUILT model then iteratively proceeds between Gibbs sampling, to obtain new read labels given the current subset of reference haplotypes, and full haploid imputation, to obtain new reference haplotype subsets using the current read labels. QUILT completes after a prespecified number of iterations. Genotype dosage is taken as an average across Gibbs samplings, while phase is taken from an additional Gibbs sampling using read labels taken as an average across previous samplings.
对NA12878的填充性能
通过standard Illumina short-read (Illumina)、haplotagged Illumina short-read (HT)和long-read (ONT) sequences,作者获得了NA12878的高覆盖率序列。通过这三份数据和一个单倍型参考群体,作者推断了准确的基因型和单倍型,并将其作为真实单倍型。接着,作者填充了基因组上200Mbp的长度作为对比。参考群体取自HRC 数据集[2],并删掉了其中的NA12878记录。后续小节中,作者采用去除exact matches的不同HRC子集来进行测试。
作者通过可视化比较读段标签从推断值到真实值的连续性,和相位切换错误率(phase switch error rate)来评估定相精度。随着算法的进行,QUILT可以得到愈发精准的读段标签,最终以约每Mb发生一次转换错误的水平构建读段水平的phasing(图2,绘制了在染色体20上的连续20Mb区间,错误可以用不同runs间蓝色和橙色的翻转来可视化)。
跨平台比较表明,standard short-read Illumina数据相较于longer-read HT or ONT产生了更多切换。Indeed phase switch error rates were 0.09% for ONT, 0.08% for HT and 0.13% for Illumina at 1.0× coverage。不同覆盖率数据定相准确率非常高。
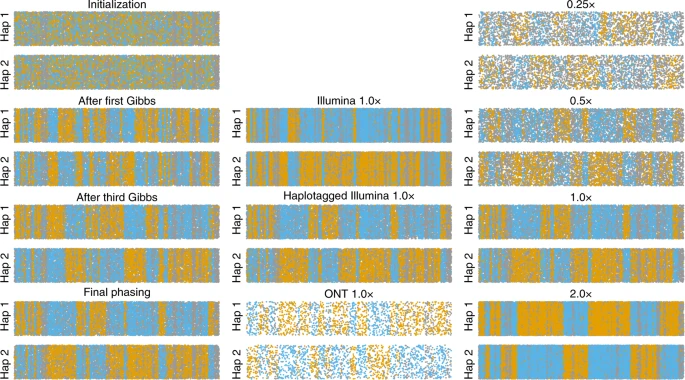
Fig. 2: Assessment of read label partitioning.
Per analysis, reads were grouped based on assignment to Hap1 or Hap2, with the remaining y axis variation being jitter. The x axis gives the central location of the read along 20 Mbp of chromosome 20. Reads are colored blue and orange to reflect a high posterior probability of coming from a truth maternal or paternal chromosome, while gray indicates equally likely from either truth chromosome. Switches between runs of orange and blue denote probable switch errors. The columns denote the effect of multiple iterations (leftmost for haplotagged 1.0×), different technologies (center, for 1.0×) and coverages (rightmost for haplotagged).
作者接着评估了跨数据类型、方法和不同覆盖率的基因型填充准确性。为了探索定相错误对基因分型错误的贡献,作者向 QUILT 框架提供了包含“真实”读段标签的ref panel,以生成所谓的“最佳”填充结果。作者采用the separate Genome Aggregation Database (gnomAD)[3]中的等位基因频率对结果进行分层。在结果中,用“rare”表示等位基因频率在0.1-0.2%的SNP,而“common”代表20-50%的SNP。令人振奋的是,0.5x的填充结果非常接近最优结果,并展示了HT测序方法的优越性(尤其在稀有SNP中)。在更低覆盖率如0.1x中,HT的优越性依然存在。
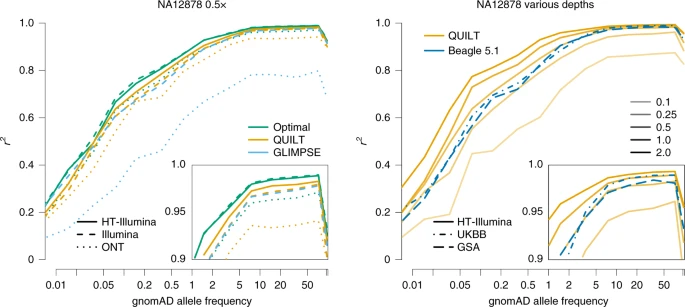
Fig. 3: Imputation accuracy of the NA12878 sample.
The \(r^2\) per bin was aggregated over SNPs with a given gnomAD allele frequency for a given technology, coverage and method.
在与GLIMPSE软件的比较(图3a)中,由于GLIMPSE采用的输入格式为VCF,其在使用linked-read HT数据时相较于standard Illumina sequencing并无优势。在0.5x下,QUILT 和 GLIMPSE 的结果相似,QUILT HT 最准确。对于 ONT,GLIMPSE 的准确度比 QUILT 低得多。在不同参考群体大小下运行速度对比中,小型(n=3)和中型(n=93)数据集下,QUILT都显出了近似线性的计算复杂度,而GLIMPSE在中等大小下显出了常数计算复杂度。两种方法在内存使用和参考群体大小之间都具有低且近似线性的关系。
QUILT 和 GLIMPSE 都包括权衡精度与运行时间的参数设置。在不同参数范围下运行两种软件,结果表明(在考虑速度和准确性时),对于覆盖率较低的数据 (0.1×),对于所有三种数据类型,尤其是 HT 和 ONT 数据,QUILT 优于 GLIMPSE。对于中等覆盖率(0.5×),除ONT 外,两方法类似;对于高覆盖率数据(1.0×、2.0×),对于 Illumina 和 HT 数据类型,GLIMPSE 优于 QUILT,而对于 ONT 数据,QUILT 显然更优。
QUILT与 genotyping microarrays的比较中,模拟得到的Affymetrix UK Biobank (UKBB)和Illumina Global Screening Array (GSA)芯片的数据利用 Beagle v.5.1 进行填充(图3b)。两种芯片在rare和common SNP上的准确性相似。对于rare SNP,所有三种方法在1.0x覆盖率下就能得到芯片的准确率。对于表现最好的HT测序平台,甚至0.5x下就能得到与芯片一致的准确率。
不同测序样本的填充性能
在另外三个数据集中,作者确认了之前在NA12878中发现的数据类型和填充性能的整体模式。
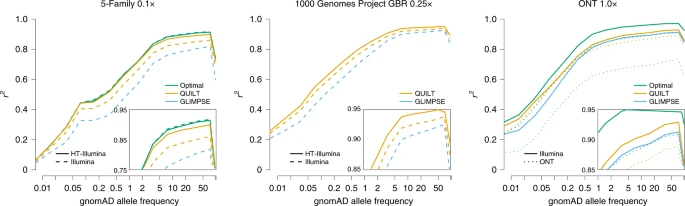
Fig. 4: Imputation accuracy of 5-Family, GBR and ONT samples.
The \(r^2\) per bin was aggregated over all SNPs in that gnomAD allele frequency bin across all samples, for a given technology, coverage and method.
相对于基因分型阵列的插补性能
接下来,作者使用 QUILT 1000 基因组计划的样本评估了来自不同地区群体的五个分组(ASW、CEU、CHB、PJL 和 PUR)的不同样本的填充性能与基因分型微阵列(UKBB 和 GSA)的比较。正如预期,CEU(北欧和西欧血统)样本(图5)的填充准确度最高(它们与大部分 HRC 参考群体最为相似),并且对于稀有 SNP,芯片的填充精度可对标 0.25 × 测序数据使用 QUILT 填充的结果。有趣的是,对于其他组,尽管绝对插补质量下降,但与基因分型芯片相比,low-coverage whole-genome sequencing (lcWGS) 和 QUILT 的相对表现更好。因此,对于与参考数据集不太相似的群体,测序比起基因分型的好处似乎要大得多。更高覆盖率的 1.0x 和 2.0x lcWGS 数据在所有频率上都优于芯片,尤其是低频变异,QUILT 几乎是上述基于阵列的值的两倍。
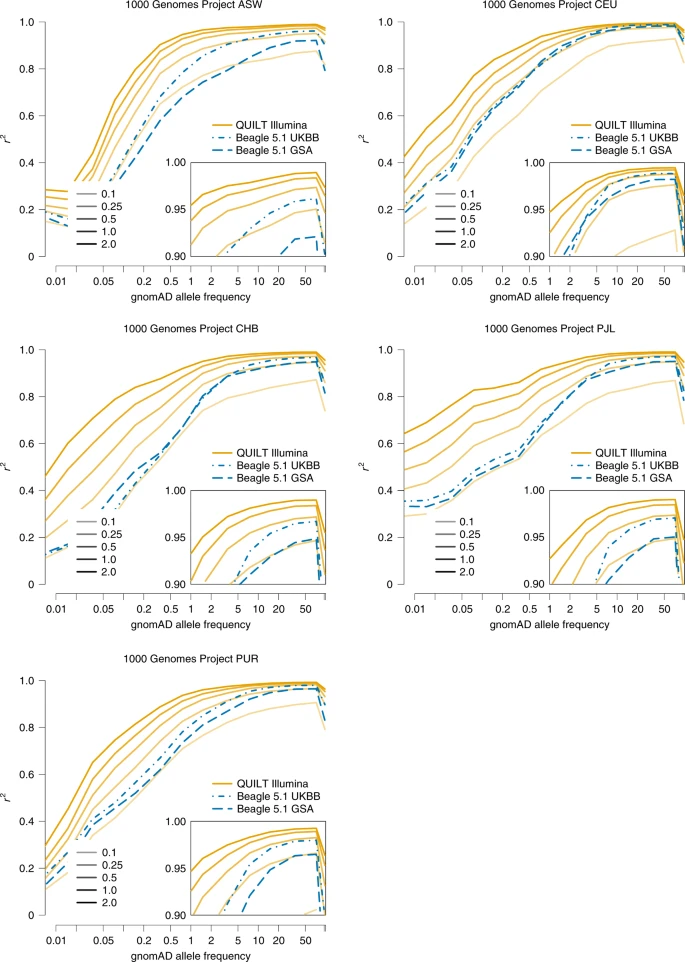
Fig. 5: Imputation accuracy of the 1000 Genomes Project samples.
The \(r^2\) per bin was aggregated over all SNPs in that gnomAD allele frequency bin across all samples, for a given technology, coverage and method.
HLA 填充性能
利用基因分型或高覆盖率测序数据进行准确的human leukocyte antigen (HLA)填充的方法已被开发出来。作为QUILT的一部分,作者为lcWGS开发了一个新的HLA填充算法,利用位于HLA locus内部的读段进行直接比对,并用其余的读段进行基于打过标签的参考群体的填充。作者采用的参考HLA数据集来自HLA database IPD-IMGT/HLA和 1000 Genomes Project,并使用保留的 1000 Genomes Project 数据进行测试。作者在五个经典 HLA 位点(HLA-A、HLA-B、HLA-C、HLA-DQB1、HLA-DRB1)上估计了五个群体(ASW、CEU、CHB、PJL 和 PUR)的 HLA 类型。作者还用SNP2HLA软件评估了利用相同参考群体的、基于芯片的HLA填充方法。
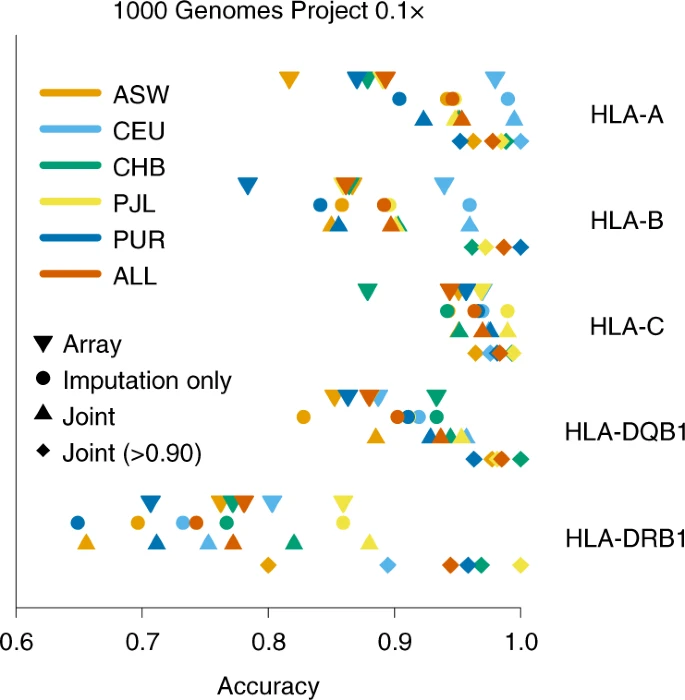
Fig. 6: Imputation accuracy of HLA loci.
Accuracy is the percentage of correct unphased HLA alleles versus computationally inferred truth. Results are shown both per population and in aggregate (ALL). Results are given both using only imputation as well as imputation plus direct read mapping (joint, the default QUILT output). Results are further given at the subset of individuals with confidently inferred alleles (joint (>0.90)). As reported elsewhere, HLA class I loci (HLA-A, HLA-B and HLA-C) are less diverse than class II loci (HLA-DRB1 and HLA-DQB1) and thus yield more accurate imputation results.
相对有效样本量和功效
最后,作者对 lcWGS 加基于 QUILT 的插补与广泛使用的基因分型微阵列进行了成本效益分析。作者假设a fixed cost of £30 per array and library costs from Meier et al.,并认为表型测性和每一倍覆盖率的价格可以从现在的US$1,000/30× genome降低到未来的US$250/30× genome。
结果表明,对于这两种设置,测序产生的有效样本量几乎一致,比基因分型阵列产生的有效样本更大(图7)。对于 GWAS 测试,lcWGS + QUILT 产生的有效样本大小分别是使用基因分型微阵列的 3.5 倍和 1.4 倍(分别以廉价和昂贵的表型成本来识别与 0.1-0.2% 频率的稀有 SNP 的关联)。
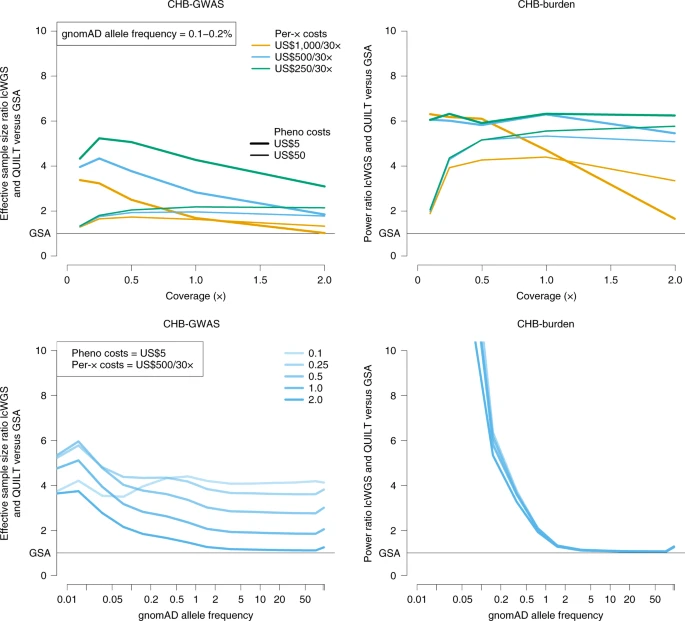
Fig. 7: Relative increase in effective sample size and power using lcWGS and QUILT.
Results are shown as a ratio of effective sample size for the GWAS setting and a ratio of power for the burden test setting. Results use the 1000 Genomes Project CHB imputation accuracy. Top, Results are given as a function of coverage, with variable phenotyping and per-× sequencing costs, for a fixed allele frequency (0.1–0.2%). Bottom, Results are given as a function of allele frequency, with varying coverage, assuming fixed phenotyping (US$5 per sample) and per-× sequencing costs (US$500 per 30×). All results assume a library preparation cost of £1.36 per sample and an array cost of £30 per sample.
其他关键参考文献
Li, N. & Stephens, M. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics 165, 2213–2233 (2003). ↩︎
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016). ↩︎
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). ↩︎

 浙公网安备 33010602011771号
浙公网安备 33010602011771号