文献阅读 | Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes
Krogh, A., Larsson, B., Von Heijne, G., & Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. Journal of molecular biology, 305(3), 567-580. https://www.sciencedirect.com/science/article/pii/S0022283600943158
在这篇2001年的文章中,作者采用了HMM模型,对跨膜蛋白(transmembrane protein)的拓扑(topology)结构进行了预测。
背景
完整膜蛋白中跨膜螺旋的预测是生物信息学的重要方面。迄今为止,最成功的方法不仅可以预测单个跨膜螺旋,而且可以尝试预测蛋白质的完整拓扑,即跨膜螺旋的总数及其相对于膜的入/出方向(von Heijne,1999)。用于区分膜蛋白和可溶性蛋白以及用于拓扑预测的可靠方法在基因组分析中具有重要的应用,并可用于提取膜蛋白进化的整体趋势(Wallin&von Heijne,1998)。
预测跨膜螺旋的早期方法仅使用疏水性分析(e.g. Argos et al., 1982)。根据疏水性图,可以高可靠性地定位某些螺旋,但其他一些则不能。
另一个与跨膜螺旋相关的信号,是膜的细胞质一侧序列中部分带正电荷残基的丰度即“正内部规则”(the positive inside rule;von Heijne 1986, von Heijne 1994)。通过将电荷偏差分析与疏水性分析相结合,可以获得更好的预测(von Heijne,1992)。尽管它们在细节上有所不同,但是几乎所有用于跨膜螺旋预测的最新方法都依赖于这两个信号。
一些方法使用滑窗法,通过权重矩阵(Edelman,1993)或通过神经网络(Rost et al 1995, Casadio et al 1996)来预测是否为膜螺旋的一部分。一些方法使用多重比对来改善预测效果(Persson and Argos 1994, Rost et al 1996)。
螺旋膜蛋白遵循一种“语法”,其中细胞质环和非细胞质环必须交替。语法限制了可能的拓扑,从而限制了可能的跨膜螺旋。因此,即使在单个跨膜螺旋预测的水平上,原则上考虑语法的集成预测方法也可以给出更好的结果。Jones et al. (1994) 描述了一种动态编程算法,该算法可以使残差得分的总和最大化,同时遵循语法。对螺旋中间,螺旋帽和环区域中的残基使用不同的分数。该方法以一种自然的方式将疏水信号和电荷偏置信号隐式组合为一个集成算法。
在HMM方法中,另一种HMM方法HMMTOP已被独立开发(Tusnady & Simon, 1998)。它以非常相似的HMM架构为基础,但是用于预测的方法却有所不同。它从一组已知的跨膜蛋白中估计出一个模型正则化器,并且为了进行预测,从查询序列中估计出一个模型,然后将其用于预测该序列的结构。HMMTOP报道的单序列预测准确度(正确拓扑为78%)与TMHMM大致相同,尽管由于数据集和交叉验证方法的差异,对精度进行对比比较困难。
TMHMM
对膜蛋白的各个区域进行了专门建模:螺旋帽(helix caps),螺旋中部(middle of helix),靠近膜的区域(regions close to the membrane)以及球状结构域(globular domains)。

图1(a)展示了模型的结构(layout),图中的每个方框对应于设计为对膜蛋白的特定区域建模的子模型。这些子模型包含几个HMM状态,以便对各个区域的长度进行建模。箭头显示了如何在子模型之间进行过渡,以使其遵循螺旋跨膜蛋白的语法结构。
作者没有尝试构建跨膜蛋白球状结构域的复杂模型,因此图1a中标记为“球状”的子模型是相同的,并且仅包含一个状态,该状态向其自身和循环转移(图1b)。为了捕获蛋白质的拓扑信号,作者在标记为“环”和“帽”的子模型中对靠近膜的残基进行建模。长度高达20个残基的循环由循环模型建模,而更长的循环则必须使用球状状态。拓扑状态的转移可确保允许长度为一个或多个的任何循环。循环子模型的20个回路状态均具有相同的氨基酸残基分布,但三个回路模型不同。cap子模型仅对跨膜区域的五个第一个或最后一个残基进行建模。

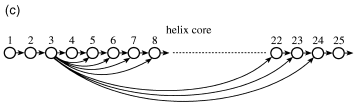
跨膜螺旋核心的模型如图1c所示。它由25个相同的状态组成,有可能从某一状态(如图中的状态3)跳到下游的许多状态。此拓扑对长度在5到25之间的序列进行建模。当包括cap时,这将转换为15到35之间的螺旋长度。这与已知结构的膜蛋白中螺旋长度的分布是一致的(Bowie,1997)。
上述模型中,长度分布由螺旋模型中过渡的过渡概率明确表示。HMM参数(是隐状态中20个氨基酸残基的概率和确定跨膜螺旋等的长度分布的概率)是从一组已知跨膜螺旋位置的160种蛋白质中估算出来的。螺旋的边界通常是不准确地确定的(甚至是从晶体结构确定的),因此作者设计了还一种估计程序,其中使用模型来重新定义边界。
跨膜螺旋的预测是通过在给定HMM的情况下找到最可能的拓扑来完成的。这将给出一组确切的螺旋边界。但是,存在许多几乎相同的可能方式来放置螺旋边界,并且序列中的某些区域有时显示出较弱的迹象,即跨膜螺旋或预测螺旋的可能性很低。此类信息未包含在最可能的预测中。因此,作者发现使用给定残基在跨膜螺旋中,在细胞质侧或在周质侧的三个概率也非常有用。该附加的信息,其可以用图形显示为在图2中,示出了其中所述预测是某些和什么替代可能存在。

图2
单个序列的后验概率。大肠杆菌中葡萄糖酸通透酶3的跨膜螺旋、膜内部或膜外部的后验概率图。该酶结构未知。蛋白质的某些部分相对确定,而其他部分则不太确定。目前尚不清楚,例如在氨基酸100和150之间以及在325和375之间存在一个还是两个跨膜片段。这种不确定性还反映在一个总的不确定性中,即环的150-325之间,究竟在膜的那一侧。
对于这种蛋白质,单个最可能的拓扑原来在这两个区域中都有两个螺旋,即共有13个跨膜螺旋,并且该预测与SWISS-PROT中的注释基本相同。但是,后验概率图显示,在这些区域中只有一个螺旋(总共11个)的拓扑结构是一个很可能的替代方案,而具有12个或14个跨膜螺旋结构的拓扑结构不太可能,因为它与后验概率很不匹配在蛋白质两端的内部/外部。Klemm et al. (1996) 预测,该蛋白有14个跨膜螺旋。预计在100到150之间的区域会出现三个螺旋。
错误的预测
当预测膜蛋白的拓扑结构时,可能会发生几种类型的错误预测。最简单的错误是过度预测和预测不足,即预测一个不存在或缺少真实跨膜区域的跨膜区域。另一类错误是将两个相邻的跨膜区域连接在一起,因此将它们预测为单个长区域,作者将其称为“错误合并”(false merge)。类似地,长跨膜区域可能被错误地预测为两个短区域,在这里被称为“错误分裂”(false split)。当然,可以正确地预测所有螺旋,但是可以将整体拓扑预测为真实拓扑的逆,即倒置拓扑。如果预测的跨膜螺旋与真实螺旋至少重叠五个残基,则认为该螺旋正确。如果失败,则认为至少有一个氨基酸与真实螺旋重叠的位移预测。已知信号肽未从序列中除去,也未计为有效的跨膜螺旋。信号肽有时很难与跨膜螺旋区分开,如下所述。
交叉验证实验重复了40次,并计算平均值和标准偏差。这些数据显示在表1中。

非膜和膜蛋白的区分
作者的这一方法也可用于区分螺旋膜蛋白和其他蛋白。

图3
跨膜螺旋的预期数目与预测的螺旋数目之间的相关性。插入图显示在0.9个预期的跨膜螺旋附近的临界区域数量爆炸,其中正点向上移动0.02,以分离正负。请注意,负数集的点表示交叉验证模型的平均值,因此,预测的螺旋数不一定是整数。误差线显示了十个模型的标准偏差。

图4
跨膜螺旋的预期数目与跨膜螺旋的预期氨基酸数目之间的相关性。仅显示了大约0.9个预期跨膜螺旋的临界区域周围的点。负点上的误差线显示了十个交叉验证模型的标准偏差。
通过该部分研究,作者以假阳性率极低的方法,从数据集中识别all but one transmembrane protein。

图5
跨膜蛋白和可溶性蛋白的区别。根据跨膜螺旋中预期的残基数进行区分。假阴性(连续线)和假阳性预测(虚线)的分数作为截止值的函数。虚线在18,这是作者使用的截止点。
信号肽和孔蛋白
靶向蛋白质输出的信号肽包含一个疏水区域,该区域很容易被预测程序误认为是跨膜区域。
对于真核和革兰氏阴性细菌信号肽,TMHMM错误地将约20%识别为跨膜螺旋。但是,对于来自革兰氏阳性细菌的信号肽,预计将有60%是跨膜螺旋。据推测,这是因为来自革兰氏阳性细菌的信号肽比其他两类具有明显更长的疏水区(von Heijne&Abrahmsen,1989)。

全基因组膜蛋白分析
考虑到TMHMM在区分膜蛋白和可溶性蛋白以及拓扑预测方面明显优于预测程序TOPPRED和ALOM(von Heijne 1992, Klein et al 1985),因此作者重复了较早的分析(Wallin和von Heijne ,1998),即在具有完整测序基因组的生物体中寻找膜蛋白。现在,我们可以更好地估计每种生物中的膜蛋白数量,还可以更好地估计在不同生物中发现不同拓扑结构蛋白的频率。
作者从训练集中的所有160个序列中训练了一个模型。确定该模型在阴性组上获得的假阳性数与上述结果一致后,将该模型用于基因组研究。
对于每个基因,计算跨膜螺旋中预期的氨基酸残基数以及拓扑预测。
信号肽有时被错误地预测为跨膜螺旋。为了纠正这个问题,作者在N末端选择了带有预测跨膜螺旋的所有蛋白质,这可能是信号肽。这些蛋白用SignalP-HMM分析(Nielsen&Krogh,1998),如果预测到了信号肽,则将其从蛋白质中去除。对SignalP-HMM准确性的初步测试表明,发现了约80%的真实信号肽,而20%的跨膜螺旋被误认为是真核生物中的信号肽。
HMM训练
作者分了第三部进行HMM参数的估算。
第一阶段中,为了校正带注释的跨膜螺旋的不正确边界,作者允许边界周围的六个残基与模型中的任何状态匹配。通过在蛋白螺旋的细胞质边界处的残疾标签添加通配符标签,采用Baum-Welch算法进行参数的估计。
在第二阶段,采用上步模型重新估算了螺旋边界。这是通过再次“取消标记”螺旋边界(这次是在每个侧面有五个残基),然后找到受剩余标记约束的最可能的标记来完成的。这意味着该蛋白质的整体拓扑是固定的,但是该模型决定了将螺旋边界放在十个残基的窗口内的位置。重新标记后,估计了一个新模型,其中所有标签均已固定。此估计从第一阶段的模型开始,并且使用Baum-Welch程序进行了估计,且没有噪声。
第三阶段和最后阶段,采用了Sonnhammer et al. (1998)的描述性判别方法进一步优化了第二阶段的模型

 浙公网安备 33010602011771号
浙公网安备 33010602011771号