文献阅读 | SSRgenotyper: A simple sequence repeat genotyping application for whole‐genome resequencing and reduced representational sequencing projects
Lewis DH, Jarvis DE, Maughan PJ. SSRgenotyper: A simple sequence repeat genotyping application for whole-genome resequencing and reduced representational sequencing projects. Appl Plant Sci. 2020 Dec 3;8(12):e11402. doi: https://doi.org/10.1002/aps3.11402.
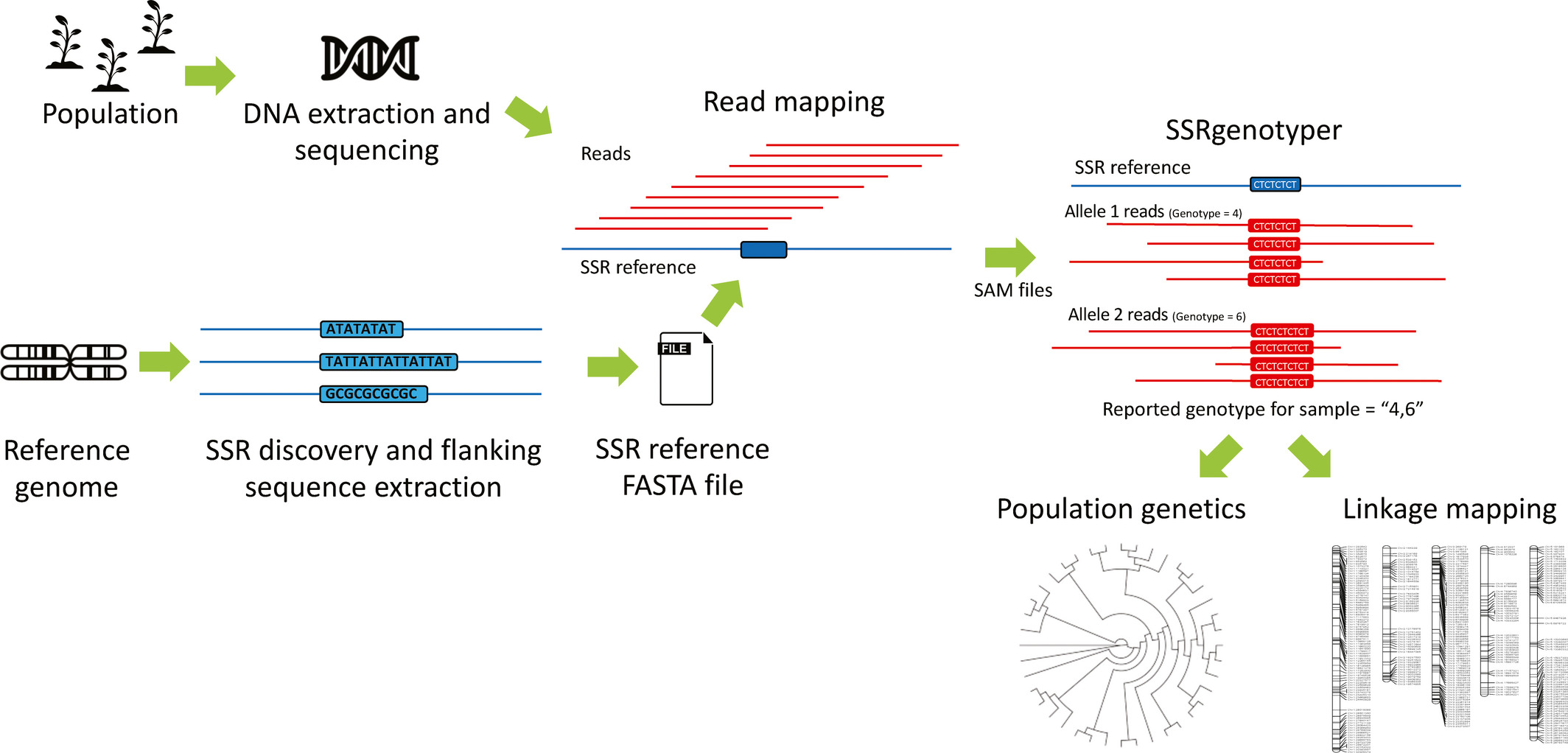
本文作者开发了一款从重测序(下机)数据对SSR进行基因分型的SSRgenotyper。
SSR标记
SSR简单序列重复标记(Simple sequence repeat, 简称SSR标记),也叫微卫星序列重复,是由一类由几个核苷酸(1-5个)为重复单位组成的长达几十个核苷酸的重复序列,长度较短,广泛分布在染色体上。由于重复单位的次数的不同或重复程度的不完全相同,造成了SSR长度的高度变异性,由此而产生SSR标记或SSLP标记。虽然SSR在基因组上的位置不尽相同,但是其两端序列多是保守的单拷贝序列,因此可以用微卫星区域特定顺序设计成对引物,通过PCR技术,经聚丙烯酰胺凝胶电泳,即可显示SSR位点在不同个体间的多态性。
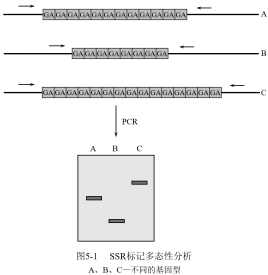
SSR标记以其多态性丰富、提供遗传信息多、操作便利并在基因组中分散分布等优点已成为最受人们欢迎的分子标记之一,在许多领域广泛应用。但SSR标记的主要缺点是首先要从该物种中获取重复序列两侧的序列信息,并设计引物,而后才能被利用。[1]
作者提到,SSR通常被认为是选择性中性的,主要受基因流、遗传漂移和突变的影响,使其特别适用于群体遗传研究。SSR也已广泛用于鉴定数量性状基因座,特别是与连锁图谱构建相结合时。
传统上,使用PCR和SSR序列保守侧翼区域的引物确定SSR基因型,然后使用高分辨率凝胶电泳或毛细管电泳仪观察共扩增产物。凝胶电泳方法很繁琐,需要超细琼脂糖(例如MetaPhor琼脂糖)或聚丙烯酰胺凝胶,且电泳时间长,然后用插层剂(通常是溴化乙锭)染色。聚丙烯酰胺和溴化乙锭都对人类健康构成威胁。另外,也可以使用专门的片段分析毛细管测序仪从荧光团标记的PCR产物中确定SSR基因型。尽管荧光团标记的引物更安全,它们昂贵得多,并且需要大量资本设备投资。荧光团标记的SSR可以多重使用以降低成本;然而,多路复用方案需要大量的开发时间,并且容量有限,通常允许少于9个SSR。此外,由于协议和仪器的差异,引物序列的变化以及缺乏标准化的调用方法,导致固有的实验室间差异,使SSR基因分型变得复杂,这些都阻碍了将数据从一个实验转移到下一个实验的能力。
SSRgenotyper
该程序旨在从大型群体的重测序数据中进行SSR分型。
其输入文件为:SAM文件和SSR参考FASTA文件
其输出内容为:SSR标记名称、位置、SSR基因型的简单表格,由repeat motif的repeat number定义
检测原理:SSRgenotyper直接从测序reads中call SSR genotypes。该程序将从序列比对/图谱(SAM)文件和SSR参考FASTA文件中找到基于二核苷酸,三核苷酸和四核苷酸的SSR。单核苷酸SSR像SSR参考FASTA文件中的软掩膜序列一样被排除在分析之外。
SSRgenotyper仅在显示正常二体遗传的二倍体和同种多倍体生物上进行过测试。由于当前由SSRgenotyper在SSR基因座处检测到的最大等位基因数目限制为两个,因此应谨慎使用SSRgenotyper来显示非二倍体遗传(例如三体性,四体性等)和自体多倍体。
SSR参考FASTA
SSRgenotyper需要一个SSR参考FASTA文件,该文件提供要进行基因分型的每个目标SSR的序列信息,包括100 bp的上游和下游侧翼序列。SSR参考FASTA文件可以包含从先前的SSR发现项目先验获得的SSR序列信息,或作为目标物种(通常是数以万计的推定SSR基因座)中发现的所有SSR的全面表示。如果目标物种有可用的基因组组装,则可以使用MISA,BEDTools和几个shell命令轻松创建全面的SSR参考FASTA文件。
附录1中提供了从参考基因组生成全面的SSR参考FASTA文件的分步指导。简而言之,MISA用于生成通用特征格式(GFF)文件,其中包含参考基因组组件中SSR基因座的位置。参考基因组可以是金标准,染色体规模的参考基因组,也可以是包含数千个重叠群的简单草图。Shell命令用于删除复合SSR和侧翼序列不足(<100 bp;重叠群边缘的序列)的任何基因座。然后,使用BEDTools提取SSR基因座的序列(包括100 bp的侧翼序列),以形成全面的SSR参考FASTA文件。
读段映射
将总体中每个个体的测序读数映射到SSR参考FASTA文件,以为每个个体生成SAM文件。SSRgenotyper不需要特定的读物类型(技术或长度),仅要求读物应高度准确才能保持基因分型质量。
读段应先进行质量控制和修剪(例如Trimmomatic),然后再映射(例如BWA-MEM ,minimap2 和bowtie2)到SSR参考FASTA文件。
由于SSRgenotyper基于从映射读段中识别的等位基因比率来进行等位基因调用,因此重要的是从SAM文件中删除PCR重复读段和任何映射不佳的读段。作者在附录2中提供了使用BWA-MEM进行Illumina配对末端读取的读取映射和处理所得SAM文件的逐步详细信息,包括用于处理大量群体中的多个个体的简单循环命令。
SSR基因分型方法
用Python 3编码的SSRgenotyper需要三个位置参数,特别是SSR参考FASTA文件(附录1),一个文本文件,其中列出了群体中每个个体的SAM文件的名称(附录2,步骤5),以及结果的输出名称。SSR基因分型:
(1)使用用户指定的边界核苷酸识别SSR重复序列的边界
(2)验证SSR与参考重复序列是否完美匹配(删除任何不匹配的读段;仅分析完美重复序列
(3)计算重复序列的总核苷酸长度
(4)通过将重复序列长度除以核苷酸基序类型(二核苷酸为两个,三核苷酸为三或四核苷酸重复为四)来确定等位基因。
因此,等位基因是基于重复的数目,而不是基于PCR产物的大小,从而使其更易于在实验和实验室之间转移。SAM文件的处理速度快,内存占用率最低,并且可以在廉价的台式计算机上完成。
附录1:基于基因组组装的SSR参考FASTA生成流程
# 1. Run MISA:
perl misa.pl my_Reference.fasta
#MISA requires a misa.ini file in the directory where MISA is being executed that should look like this:
#definition(unit_size,min_repeats): 2‐6 3‐4 4‐4
#interruptions(max_difference_between_2_SSRs): 100
#GFF: true
# 2. Modify the MISA‐produced .gff files as follows:
#A. Remove any compound SSRs and calculate how much flanking sequence is available at each SSR locus:
for i in *.gff; do grep ‐v "compound" $i | awk '{if ($5‐$4 >10 && $5‐$4 <50) print $1 "\t" $4‐100 "\t" $5+100}' > $i.mod.gff; echo "processing $i"; done
#We note that the size of the flanking region can be changed using the awk statement above (simply change the “‐100” and “+100” to the flanking size wanted).
#B. Concatenate the modified .gff files:
for i in *.mod.gff; do cat $i >> cat.gff; echo "processing $i"; done
#C. Remove SSRs that do not have sufficient flanking sequence:
awk '($2 >= 0)' cat.gff > cat_filter1.gff
#D. Use BEDTools getfasta to make the SsrReferenceFile.fasta:
bedtools getfasta ‐fi my_Reference.fasta ‐bed cat_filter1.gff ‐fo SsrReferenceFile.fasta
附录2:使用SAMtools进行BWA-MEM读段映射、PCR重复读段去除和读段质量控制的流程
# 1. Index the SsrReferenceFile.fasta:
bwa index SsrReferenceFile.fasta
# 2. Map the Illumina reads to the SsrReferenceFile.fasta (paired‐end reads process shown):
for forward_file in *_1P.fq.gz; do name=echo $forward_file | sed 's/_1P.fq.gz//'; bwa mem ‐M ../reference/ SsrReferenceFile.fasta ${name}_1P.fq.gz ${name}_2P.fq.gz ‐o $name.sam; done
#Note: Raw reads should be pre‐trimmed. Using Trimmomatic (Bolger et al., 2014) will produce a *_1P.fq.gz and *_2P.fq.gz file for each sample.
# 3. Remove PCR duplicate reads:
# A. Sort SAM files by read name
for i in *.sam; do samtools sort ‐n ‐o $i.sorted $i; done
# B. Identify mate coordinates
for i in *.sorted; do samtools fixmate ‐m $i $i.fixmate; done
# C. Re‐sort SAM files by genomic location
for i in *.fixmate; do samtools sort ‐o $i.position $i; done
# D. Mark and remove duplicates:
for i in *.position; do samtools markdup ‐r $i $i.markdup; done
#*Each sample will have a markdup file where PCR duplicates have been removed (the other files can be deleted).
# 4. Read mapping quality control:
# A. While the whole SAM file can be passed to SSRgenotyper, we encourage users to first filter the markdup file with SAMtools to improve performance and to remove errantly mapped reads. This command will remove reads with mapping quality < 45.
For i in *.markdup; do SAMtools view $i ‐q 45 > $i.Q45.sam; done
#Note: SSRgenotyper provides further filtering (option ‐Q) that can be used for additional filter stringency if required.
# 5. A .txt file listing all of the quality‐controlled SAM files (SamFiles.txt) to be processed by SSRgenotyper can be produced with:
ls *.Q45.sam > samFiles.txt
附录3:测试中使用的命令,含BioProject号和SRA标识符。
# 1. Run command for accuracy and error profile analysis, where ‐S was provided at various read coverage depths, ranging from 1 to 10 (C. quinoa: PRJNA640095, SRA no.: SRR12041554–SRR12041555; A. atlantica: PRJNA546592):
python3 SSRgenotyperV3.py SsrReferenceFile.fasta samFiles.txt quinoa ‐M 0.35 ‐R 4 ‐P 3 ‐B 3 ‐S # ‐F 0.35 ‐f 0.30 ‐Q 60 ‐s 0.1 ‐m 0 ‐N 11
# 2. Run command for the Arabidopsis thaliana recombinant inbred line (RIL) population (BioProject no.: PRJNA418075, SRA no.: SRR6292944–SRR6293128; Serin et al., 2017):
python3 SSRgenotyperV3.py SsrReferenceFile.fasta samFiles.txt BayreuthXShahdara ‐M 0.51 ‐R 4 ‐P 3 ‐B 3 ‐S 1 ‐F 0.35 ‐f 0.30 ‐Q 60 ‐s 0.1 ‐m 0 ‐N 11 ‐L 0.25
# 3. Run command for the foxtail millet RIL population (BioProject no.: PRJNA562988, SRA no.: SRR10038747–SRR10038795; Liu et al., 2020):
python3 SSRgenotyperV3.py SsrReferenceFile.fasta samFiles.txt foxtail ‐M 0.51 ‐R 4 ‐P 3 ‐B 3 ‐S 1 ‐F 0.35 ‐f 0.30 ‐Q 60 ‐s 0.1 ‐m 0 ‐N 11 ‐L 0.25
# 4. Run command for the quinoa diversity panel (BioProject no.: PRJNA306026, SRA no.: SRR4300210–SRR4300229; Jarvis et al., 2017):
python3 SSRgenotyperV3.py SsrReferenceFile.fasta samFiles.txt quinoa ‐M 0.25 ‐R 4 ‐P 3 ‐B 3 ‐S 4 ‐F 0.35 ‐f 0.30 ‐Q 60 ‐s 0.1 ‐m 0 ‐N 11 ‐G
其他参考文献
张增翠,侯喜林. SSR分子标记开发策略及评价[J]. 遗传, 2004, 26(5): 763-768. ↩︎

 浙公网安备 33010602011771号
浙公网安备 33010602011771号