

jucer和3d-DNA及其文章
Juicer allows users without a computational background to transform raw sequence data into normalized contact maps with one click.
Juicer produces a hic file containing compressed contact matrices at many resolutions, facilitating visualization and analysis at multiple scales.
Juicer comprises three tools, which are designed to be run one-after-another.
First, Juicer transforms raw sequence data into a list of Hi-C contacts (pairs of genomic positions that were adjacent to each other in three-dimensional space during the experiment).
To accomplish this, read pairs are aligned to the genome; both duplicates and near-duplicates are removed, and read pairs that align to three or more locations are set aside.
When appropriate hardware is available, this procedure can be accelerated, either by parallelizing across multiple CPUs or by using an FPGA.
Next, the catalog of contacts is used to create contact matrices. To do so, the linear genome is partitioned into loci of a fixed size, or “resolution,” (e.g., 1Mb or 1Kb).
These loci correspond to the rows and columns of a contact matrix; each entry in the matrix reflects the number of contacts observed between the corresponding pair of loci during a Hi-C experiment.
Due to factors such as chromatin accessibility, certain loci are observed more frequently in Hi-C experiments. Juicer can adjust for these biases in multiple ways.
A wide array of quality statistics are also calculated, making it possible to assess the success and reliability of a given experiment before the costly deep-sequencing step.
The contact matrices generated in this way are stored efficiently in a compressed format, which is designed to facilitate all subsequent computations.
For instance, 1 terabyte of raw sequencing data is represented as an 80 gigabyte hic file containing normalized and non-normalized contact matrices at 18 different resolutions, from 2.5Mb resolution to single restriction fragment resolution for a 4-cutter restriction enzyme (~400bp). Contact matrices in the hic format can also be visualized using Juicebox, which is described in the accompanying paper.
Finally, Juicer contains a suite of algorithms that are designed to annotate contact matrices and thus identify features of genome folding. These features include loops, loop anchor motifs, and contact domains.
Loops are identified using the HiCCUPS algorithm, which searches for clusters of contact matrix entries in which the frequency of contact is enriched relative to the local background. Since there are trillions of pixels in a kilobase-resolution Hi-C map, HiCCUPS is implemented using GP-GPUs. Given CTCF and/or cohesin ChIP-Seq tracks for the same cell type, HiCCUPS can frequently use FIMO to identify the CTCF motif that serves as the anchor for each loop. We recently performed CRISPR experiments disrupting seven different CTCF motifs, each of which was identified by HiCCUPS as the anchor of one or more loops. In each case, disruption of the motif led to disruption of the corresponding loop, thus confirming the accuracy of HiCCUPS loop anchor annotations.
Contact domains are identified using a dynamic programming algorithm that relies on applying the Arrowhead transformation [Ai,i+d = (M* i,i-d − M* i,i+d)/(M* i,i-d + M* i,i+d)] to a normalized contact matrix M* . Many of these domains are associated with loops, and can be disrupted by manipulating the corresponding loop anchors.
It is frequently useful to examine the cumulative signal from a large number of putative features at once, including both loops and domains. To this end, Juicer includes an implementation of Aggregate Peak Analysis.
Juicer is an open-source project. It is available at github.com/theaidenlab/juicer as a series of packages designed for a variety of hardware configurations: either a single machine, or clusters that run LSF, Univa Grid Engine, or SLURM. In addition, Juicer is available on the cloud at Amazon Web Services.displays different performance metrics on each cluster system; the details of each setup are in the supplement text.
Once installed, Juicer can be executed using a single command, by users without informatics experience.
Sequenced read pairs (horizontal bars) are aligned to the genome in parallel. Color indicates genomic position. Read pairs aligning to more than two positions are excluded.
Those remaining are sorted by position and merged into a single list, at which point duplicate reads are removed.
The .hic file stores contact matrices at many resolutions, which can be loaded into Juicebox for visualization
3d-dna文章和算法
Dudchenko O, Batra S S, Omer A D, et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds[J]. Science, 2017, 356(6333): 92-95.
研究人员开发出了一种Hi-C数据的新算法3D de novo assembly (3D DNA) pipeline
(https://github.com/theaidenlab/3d-dna),用这一改良的Hi-C数据分析技术来组装人类基因组,发现99%的基因序列符合人类基因组的标准参照,93%的scaffolds定向均是正确的。联合已有的埃及伊蚊数据采用Hi-C数据(40X序列覆盖度)来分割,锚定,排序,定向和合并10kb以上的2534个Scaffolds。其中,鉴定了1422个Scaffolds中明显的错配。组装后的AaegL4版本基因组含有三个染色体长度的Scaffolds(长度分别为307Mb、472Mb和404Mb)占总输入序列的94%,合并剩下的3981个短的Scaffolds(N50为65kb,最长为474kb)获得最终与三条染色体对应的Scaffolds。
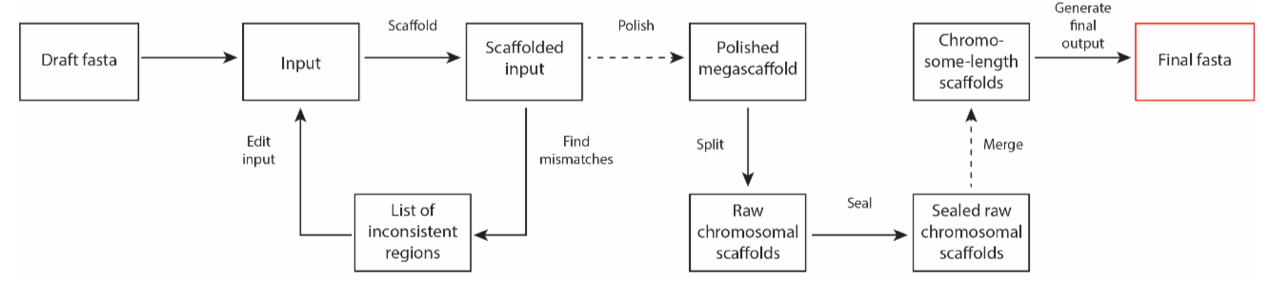
算法处理描述:
1) 首先过滤小的Scaffolds,由于其片段长度过小,Locus互作频率相对较少,分析结果不可靠。
2) 对剩余的Scaffolds进行Locus互作频率一致性分析,具有错误拼接的Scaffolds分割成段,分割后保留具有远程互作模式一致性的Scaffolds片段,去掉不具一致性的部分。
3) 根据一对互作序列之间的互作强弱来锚定,排序和定向所得到的序列,建立初步可信的染色体长度的Scaffolds。
4) 基于序列同源性和远距离互作模式高度相性鉴定基因组重叠区域,根据重叠区来合并Scaffolds和Contig,获得最终的染色体长度的Scaffolds。这一步骤对于高杂合的基因组组装至关重要。

3D-DNA是一款简单,方便的处理Hi-C软件,可将contig提升到染色体水平。其githup网址:https://github.com/theaidenlab/3d-dna
3D-DNA流程简介
- 将Hi-C数据比对到draft.genome.fa。(利用Juicer分析Hi-C数据)
- 利用自动化流程进行纠错(misjoin),排序(order),确定正确方向(orient),最后scaffolding,得到染色体水平的组装结果(3D-DNA分析)
- Juicebox 进行人工纠错

大概流程
数据准备:
- ref文件夹: 存放draft.genome.fa
- fastq: 存放HI-C测序双端reads, 注意reads文件名的格式 保证*.R1.fastq, *.R2.fastq
++++++++++++++++++++++++正式开始+++++++++++++++++++++++++++++
一、 利用Juicer 分析HI-C数据
第一步:基因组建立索引
bwa index draft.genome.fa
第二步: 创建可能的酶切位点文件
1 python ~/software/juicer/misc/generate_site_positions.py HindIII draft.genome draft.genome.fa 2 3 # 本次使用的是 HindIII 进行酶切;选择自己所有的酶
第三步:获取每条contig的长度
1 awk 'BEGIN{OFS="\t"}{print $1, $NF}' draft.genome_HindIII.txt > draft.genome.chrom.sizes
第四步:运行juicer
注意:必须在当前目录存在fastq和ref文件夹, -z,-p,-y必须参数

1 ~/software/juicer/scripts/juicer.sh -g draft_genome -s HindIII -z ./ref/draft.genome.fa -y
./ref/draft.genome_HindIII.txt -p ./ref/draft.genome.chrom.sizes -t 8 2 3 4 5 ## 参数 6 -g: 定义一个物种名 7 -s:酶切类型, HindIII(AAGCTAGCTT), MboI(GATCGATC) , DpnII(GATCGATC), NcoI(CCATGCATGG) 8 -z : 参考基因组文件 9 -y: 限制性酶切位点可能出现位置文件 10 -p: 染色体大小文件 11 -C: 将原来的文件进行拆分,必须是4的倍数,默认是90000000, 即22.5M reads 12 -S: 和任务重运行有关,从中途的某一步开始,"merge", "dedup", "final", "postproc" 或 "early" 13 -d: juicer的目录 14 -D: juicer scripts的目录 15 -t: 线程数
结果:结果文件在aligned目录下,其中"merged_nodups.txt"就是下一步3D-DNA的输入文件之一。
二、 运行3D-DNA
使用默认参数进行3D-DNA
1 ~/software/3d-dna/run-asm-pipeline.sh ./ref/draft.genome.fa ./aligned/merged_nodups.txt
最后输出文件中,包含FINAL就是我们需要的结果。
三、 juicerbox进行手动纠错
首先下载该软件:https://github.com/aidenlab/Juicebox/wiki/Download
一般组装错误为:
- misjoin
- translocations
- inversions
- chromosome boundaries
关于该软件用法,可看该视频:https://www.bilibili.com/video/av65134634
纠错完以后,会得到genome.review.assembly用于下一步的分析
四、 再次运行3D-DNA
1 ~/software/3d-dna/run-asm-pipeline-post-review.sh -r genome.review.assembly ./ref/draft.genome.fa aligned/merged_nodups.txt
假如你不小心设置了错误的-p参数,也不是特别的要紧,因为之后在最后阶段(final) 才会遇到了下面这个报错。
Could not find chromosome sizes file for: reference/genome.chrom.size
***! Can't find inter.hic in aligned/inter_30.hic
***! Error! Either inter.hic or inter_30.hic were not created
Either inter.hic or inter_30.hic were not created. Check aligned for results
即便遇到了这个报错也不要紧,因为inter.hic 和 inter_30.hic在3d-dna流程中用不到,所以不需要解决。
如果需要解决的话,有两个解决方案,一种重新运行命令,只不过多加一个参数-S final, 就会跳过之前的比对,合并和去重步骤,直接到后面STATISTICS环节。但是这样依旧会有一些不必要的计算工作,所以另一种方法就是运行原脚本必要的代码
输入文件是从hic搭建软件3d-dna获得的**.hic文件,直接输入这个文件后就可以获得一个直接的热图。
还需要一个.assemble文件,是包含了contig位置信息的文件。输入进去之后会是很多方框,表示了这个contig与热图对应的位置,然后就可以进行染色体的确定和手动调整。具体还是看3d-dna的或者juicebox的使用说明吧。
https://github.com/aidenlab/Juicebox/wiki/Visualization
De novo assembly of the Aedes aegyptigenome using Hi-C yields chromosome-length scaffolds
原始文章阅读学习
Computational experiments with simulated input data have suggested that Hi-C should be able to produce chromosome-length scaffolds (6–8). Indeed, Hi-C has been used to improve draft genome assemblies (7, 9) and to create chromosome-length scaffolds for large genomes
大多数用短reads拼接的基因组都需要额外的数据补洞,用来完成构成真核基因组的许多染色体的完整组装。Dudchenko(杜沁珂..:))Hi-C方法测量了用于scaffold确认的在染色体内和间的接触点,加上修正和排序,以更彻底地确定基因组映射的短序列的排列。他们通过一个完整的人类基因组的生成验证了他们的方法。用已有的draft基因组和Hi-C的数据产生了染色体长度的scaffold, 短reads是67x, 这些组装表明这些物种的几乎所有基因组重组是在染色体臂的内部而不是之间,发生的。这种基因组组装方法快速,便宜,精确,适用范围广。
contiguous sequences (contigs)
Within scaffolds, adjacent contigs are often separated by a gap, which corresponds to a region that is hard to assemble from the available sequence reads (for example, because of repetitive sequences or low coverage) but that can nevertheless be spanned by using the linking information to determine the contigs at either end of the gap.
这种克隆文库常常能够提供1000倍范围的覆盖度,用这个策略,它很可能产生的scaffold的大小从1到15Mb. 但由于一些重复区域太大,很难通过可获得的克隆文库得到延展,所以获得在整个染色体延伸的scaffolds是不可能的。
Hi-C 用于测量成对位点间的接触频率-来得知基因组是如何折叠的。接触频率在很大程度上取决于成对位点,(以碱基对表示)一维(1D)距离,例如,在人类基因组中分离10kb的位点,比距离为100kb的位点的接触多8倍。按绝对值计算,从给定位点,其 10 kb 以内的位点的典型的Hi-C接触分布为 15%;其10-100 Mb 远的接触点分布为16%。
Hi-C 数据可以提供跨各种长度的连接 (links), 甚至扩展到整个染色体。然而,与来自克隆文库的成对末端reads不同,任何给定的Hi-C接触跨越未知长度,并且可能连接不同染色体上的基因位点。
Computational experiments with simulated input data have suggested that Hi-C should be able to produce chromosome-length scaffolds. Indeed, Hi-C has been used to improve draft genome assemblies and to create chromosome-length scaffolds for large genomes. 在这个过程中,HiC数据被用来将草图scaffold分配到染色体上,然后排序,然后在每个染色体上排列和定位这个草图scaffold. 不幸的是,这个结果包含大量错误,包括染色体级别的倒置和错位,融合了染色体。此类装配错误有可能是因为原始的草图装配的错误引起的。避免此类错误的一个方法,可能是用其他额外类型信息的辅助,比如更长的reads或者光学映射数据。
We therefore sought to develop a robust procedure for using Hi-C linking information to generate accurate genome assemblies with chromosome-length scaffolds. A key aspect of the approach is to first use Hi-C data to identify and correct errors in the scaffolds of the initial assembly. 用HiC连接信息来识别和矫正组装草图中的错误,简单来说,我们通过识别位置来矫正错误连接,在这些位置上,一个scaffold长度范围的互作模式突然改变了,而这种情况,在正确组装的scaffold中是可能出现的。接下来,我们调用了一对序列中的互作频率,作为在1维基因组它们的接触程度的“指示剂”,使用了一个新颖的算法来锚定、排序和定位这些结果序列。最终,我们通过识别这些成对的scaffolds,这些scffolds在长范围互作模式上表现出强烈的序列同源性和强烈的相似性,将相应于这些基因组的重叠区域的contigs和scaffolds进行合并(图1和图S1)。
图1 我们使用HiC数据,对草图组装进行错误连接矫正,scaffold,合并重叠区域,因此组装除了染色体长度的scaffolds.

这里我们展示了一个通过将HiC数据集比对到基因组上的互作矩阵,
左边是输入,右边是用我们的算法装出来的最终的基因组。互作矩阵中的像素强度表明了细胞核中的一对位点共定位是有多频繁。对应每一行每一列相应的位点用染色体图谱来表明。最左边的染色体图描绘了三个连锁群:Lnk1,2,3和一个未分配的U。
右边的染色体图,描绘了三个染色体水平的scaffold,
为了建立这个染色图,我们给最右边图里的每个arm分配了一个线性的颜色梯度,因此给每个位置都指定了一个颜色。同样的颜色用在左边图里的相应的位置上,和我们中间图:矫正过程的说明中(通过逐渐增加对比)色谱图的不连续性表明了这些图和最终最右边的图的不同。
我们用左图中Lnk1中的scaffold作为输入,来描述我们的组装算法,看括号里的supercontig 1.12。
首先,检查scaffold的错误拼接,将展现出连续HiC信号的结果片段拆分(中图,最上面那副)
然后,这个片段被用作进行scaffolding的迭代的输入
最终,只有一个其中的片段被比对到了染色体1
在许多的scaffold的附近-没有锚定到左图上的剩下的超级contig1.12被比对到了2q上,(中图,中间那一行)
最终,呈现出相似3D信号的片段被检查作为重叠序列的证据(绿色的矩形),并且被合并。(中图,下面那一行)
最终的互作图是和Rabl(可能是个基因的名字)构成是一致的,例如,着丝粒和端粒的空间聚类。
我们用从头组装的人的基因组(某细胞系测的),验证了我们的方法。这个仅使用了短illumina reads (67X) 组装成了23个染色体级别的scaffold。我们从250碱基对(bp)双末端reads,(从SRA数据库下载了SRX297987-用无PCR的方法illumina测序、60X 覆盖率的数据),用DISCOVAR从头组装,这个出来的组装本,被命名为Hs1, 包括2.82Gb的序列,在73,770 scaffolds(N50:126 kb)中分出来的contig N50:103kb
表1,我们没有尝试进一步组装每个草图中包含的微小支架。而是通过使用Hi-C组装每个草图中的其他支架,以创建巨大的,染色体长度的支架和其他小的支架。
我们然后使用了原位的HiC数据(6.7X的序列覆盖度),来提高组装草图。我们把小于15kb,N50 6.1 kb的作为小的scaffolds,并把它们放在一边。它们总共占据了测序碱基的5.4%。由于它们大小很小,所以相对Hi-C互作接触少,更难分析。我们接下来使用HiC数据来分割、锚定、排序和定位剩下的3万多个scaffolds。组装结果包含23个很大的scaffolds,长度从28.8到225.2Mb,包括了总序列的99.5%, 其余811个小scaffold, N50长度是30 kb,最大长度231 kb,占据了基因组剩余的0.5%。
我们组装的基因组与人类参考基因组进行质量比较。23个scaffolds对应了23个人类的染色体,跨越了长度的99%和染色体长度scaffold的91%,这些染色体长度的scaffolds99.7%能唯一放置到人类参考基因组正确的染色体上。对于随机选择的成对的比对到相同的染色体长度上的scaffolds的scaffolds,它的顺序是和参考基因组中99%的顺序相一致的。
这个96%的一致性,反映了HiC数据对分辨短scaffolds的好的结构提供了更少的信息。尽管如此,对于至少120 kb长的scaffolds的一致性是99%。类似地,93%的scaffolds的定位是准确的,而且大多数错误都是来源于短scaffolds的。
总之,这个染色体长度、小的scaffolds占据了参考基因组染色体长度scaffolds的97.3%?剩余的主要因为是重复序列,不能从短reads中充分组装。我们的方法进一步通过PacBio三代长reads组装出来草图得到了验证,那个装出来的草图包含了更长的contigs.
接下来,我们将我们的方法应用到了一个原来用Sanger 8X 覆盖度的reads组装出来的基因组中,这个组装草图包含了1.3 Gb的序列 (contig N50是83 kb),分散在4756个scaffolds中,scaffolds N50是1.5 Mb。
为了提高这个基因组草图的质量,我们生成出了40X覆盖度的原位的HiC数据,将2222个短于10kb的scaffolds放在一边后(占据了起始组装的碱基的1%),我们使用HiC数据来切分,锚定,定位和融合了剩下的2534个scaffolds. 明显地,我们的流程识别出了这些输入scaffolds中错误连接的scaffold的56%:1422个。
我们把我们的组装本比较到了这个埃及按蚊的基因图上,在这个遗传图上,有2006个标记,其中1826个标记能够被明确地map到右图(也就是HiC辅助组装并校正后的最终图)上。明显地,我们的组装和这个遗传图的1826个标记中的1822个一致(图2)。这里的例外(1826-1822=4?)是因为在左图(也就是使用HiC辅助前的基因组草图)的错误连接,在右图(也就是用HiC完成辅助组装后)中没有检测到。我们也观察到?和该埃及按蚊基因组物理图谱的密切一致性(图S12)。

图2 HiC辅助组装最终结果AaegL4和CpipJ3的遗传图谱的比较
我们对AaegL4和CpipJ3的遗传图谱进行比较,我们的组装结果AaegL4是和遗传图谱1826中的1822个相一致的。不一致的那4个是因为在图1左图中的基因组草图中的错误拼接,在图1右图最终HiC辅助组装且校正后的最终的基因组中没有被校正。
类似地,右图CpipJ3的遗传图谱是和另一种蚊子C. q...的遗传图谱是一致的。
我们还用我们这个方法,组装了上面提到的那另一种文字C.q...。我们产生了100X覆盖率的原位HiC数据,并且用它们提高了之前版本的基因组CpipJ2,获得了一个新的组装CpipJ3,这个新装出的基因组,有3个染色体长度的scaffolds,总共占据了起始组装的94%的序列。我们通过将HiC最终辅助组装出的基因组与已有的遗传和物理图谱比较,对新装出的该HiC辅助组装出的基因组进行了验证。
两种蚊子染色体长度scaffolds的创建,允许我们使用我们的HiC数据来创建HiC热图-展现了两个基因组中染色体基因组之间的邻近关系。明显地,三个染色体的远末端展现了两个物种中的空间聚类。两个物种都展现出了一个第二个空间聚类,包含三个位点:一个位点来自于每个染色体,主要分布在中间。这种聚类与着丝粒的空间聚集相一致,着丝粒在许多生物中都存在。总之,这个3D图适合一个已知的Rabl结构的空间组织相一致的。我们的发现也表明了每个染色体中心粒的位置和它们把每个蚊子的染色体分成两个臂(arms)。
(用HiC组装出来的基因组,可以进行进化分析。)两种蚊子的全基因组比对识别出1389个大的保守共线性区块。相似的结构在另一种蚊子C. q..中也能观察到。尽管存在密集的重组,我们在某三种蚊子中,观察到了染色体arms间相对应的序列。特别是,一个物种中大量的DNA序列在一个特殊的染色体臂上,而在另外两个物种中单个染色体臂上也发现了这些同源序列。唯一的例外是在其中冈比亚按蚊中在单个arm上的某序列,是在埃及按蚊和C. q..蚊子的两个arm上的,这和导致了埃及按蚊和C. q..蚊子共有祖先这个arm的裂开有关(图3)。这些观察结果是和细胞分析学结果一致的。

图3. 蚊子的染色体arms的内容是高度保守的
埃及按蚊,每100 kb的位点,指定一种颜色
其他物种,每100 kb的位点,指定一个对应埃及按蚊颜色的组合,用长度来衡量
总之,这些结果表明,除了上面提到的分裂事件外,三种蚊子的每个染色体臂是在约1.5-2亿年前来自它们共同祖先的一个单个的arm的,蚊子对臂内重排的偏好比哺乳动物更强
值得注意的是,果蝇的2号染色体的左臂在所有这三种蚊子中都有明显的对应物。因此,所有四个臂都起源于2.5亿年前它们的的二倍体祖先中的单个染色体臂
总的来说,我们的结果表明,将Hi-C数据整合到基因组组装中可提供一种快速,廉价的方法,以生成具有染色体长度支架的高精度从头组装。目前,哺乳动物基因组的3D从头组装总测序成本低于10,000美元,而较小的基因组则更低
重要的是要记住,这种组装仍然包含错误。例如,尽管Hi-C数据提供了覆盖长距离的广泛链接,但是当前该方法对于小邻接contigs的局部排序并不理想。可以通过对Hi-C数据进行更复杂的分析来避免这种情况。附加数据(例如长读或配对读)也可以改善结果。快速可靠地生成具有染色体长度scaffold的基因组草图将能加快许多生物的基因组分析。
参考来源:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5846465/
http://aidenlab.org/documentation.html
https://www.cnblogs.com/zhanmaomao/p/12763021.html
https://www.sohu.com/a/227585271_464200
https://www.jianshu.com/p/42f424ccb2db
https://github.com/aidenlab/Juicebox/wiki/Visualization
https://science.sciencemag.org/content/356/6333/92 Dudchenko, Olga, et al. "De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds." Science 356.6333 (2017): 92-95.
posted on 2020-07-10 15:10 BPSO_mynotes 阅读(3913) 评论(0) 收藏 举报


 浙公网安备 33010602011771号
浙公网安备 33010602011771号