
论文解读 神经退行性疾病与表观遗传衰老和人类寿命的因果关系和共同遗传病因
同学们,大家好!今天给大家介绍一篇研究性论文,神经退行性疾病与表观遗传衰老和人类寿命的因果关系和共同遗传病因,这是一篇研究共病遗传机制的文献,想了解这方面的同学们可以重点关注一下。这篇文章是2024年1月份发表在Aging Cell(IF:8.4,中科院分区:医学1区-老年医学)上的,下面我们详细看看。

在该文章中,作者使用的研究方法有:
1、工具变量选择(通过全基因组显著性和连锁不平衡聚集临界值来鉴定)
2、孟德尔随机化分析
3、敏感性分析
4、错误发现率和共定位分析
5、基因座定义
6、功能注释
下面我们详细看一下:
Causal associations and shared genetic etiology of neurodegenerative diseases with epigenetic aging and human longevity
Abstract
神经退行性疾病与表观遗传衰老和人类长寿之间的遗传关系的致病机制仍然不清楚。我们旨在检测神经退行性疾病与表观遗传衰老和人类寿命之间的因果关系和共同的遗传病因。我们获得了大规模全基因组关联研究的总结统计数据的四个衡量表观遗传年龄(格里姆年龄、表型年龄、IEAA和汉纳姆年龄)(N = 34,710),多变量寿命(健康时间、寿命和超长寿命)(N = 1,349,462),以及用于多种神经退行性疾病(N = 6618- 482,730),包括路易体痴呆(Lewy body dementia)、阿尔兹海默症(AD)、帕金森病(Parkinson's disease)、肌萎缩性侧索硬化症(amyotrophic lateral sclerosis)和多发性硬化症。主要分析使用乘法随机效应逆方差加权孟德尔随机化(MR)和条件/联合错误发现率(cond/conjFDR)方法进行。对共有基因组位点进行功能表征以获得生物学理解。有证据表明,AD患者的异常寿命比正常人少0.309年(IVW beta = −0.309,95% CI:−0.38至−0.24,p = 1.51E-19)。我们还观察到AD和GrimAge年龄加速之间存在显著的因果关系(IVW beta = −0.10,95% CI:−0.188至−0.013,p = 0.02)。在发现多基因重叠后,我们鉴定了rs78143120为AD和GrimAge年龄加速之间的共有基因座,rs 12691088为AD和异常长寿之间的共有基因座。在这些位点中,rs78143120是AD的新位点。总之,我们观察到只有AD对表观遗传衰老和人类寿命有因果影响,而其他神经退行性疾病则没有。它们之间的遗传重叠和作用方向的混合,提示了复杂的共同遗传病因和分子机制。
INTRODUCTION
神经退行性疾病和人类衰老都是时间依赖性疾病,并相互伴随的报告。已知衰老是神经退行性疾病的主要危险因素,包括阿尔茨海默病(AD)、帕金森病(PD)和路易体痴呆(LBD)。这表明神经退行性疾病与人类衰老之间可能存在遗传联系和共同的生物学机制。然而,据我们所知,很少有研究研究神经退行性疾病是否有助于衰老。因此,研究神经退行性疾病是加速还是延缓衰老既是一个挑战,也是一个激励。
在全球范围内,2016年约有4400万人患有痴呆,610万人患有PD,预计到2050年AD和PD的发病率将增加一倍以上的报告。神经变性疾病已经成为一个主要的公共卫生问题,造成了巨大的社会和经济负担的报告。此外,患有这些神经变性疾病的患者经历了对其生活质量和健康寿命的严重影响。因此,揭示神经退行性疾病与人类寿命的因果关系和共同遗传病因学,可能有助于识别处于风险中的人群,促进健康老龄化。总之,有必要研究神经退行性疾病与人类衰老和长寿之间的特定遗传关系。
老化表型已被用于评价老化水平并应用于遗传研究。我们的研究集中在表观遗传钟(GrimAge,PhenoAge,IEAA和HannumAge)和人类长寿表型,包括健康寿命,父母寿命(以下简称寿命)和异常长寿。最近的几项研究指出,表观遗传年龄的测量值(也称为表观遗传钟)是目前可用的生物学年龄的最佳预测值。表观遗传年龄可能与个体的实足年龄不一致,在生物学上比实足年龄更年轻或更老,称为表观遗传年龄加速(EAA)的报告。不同的表观遗传钟捕捉生物老化过程的不同特征。汉纳姆年龄和内在HorvathAge是“第一代”表观遗传时钟。HannumAge利用基于Illumina 450 k阵列的71个CpG位点并且对于全血样品是最佳的的报告。Intrinsic HorvathAge 是一种基于 Illumina 27k 芯片在 353 个 CpG 位点上训练的多组织年龄预测因子,可以估计大多数组织和细胞类型的 DNA 甲基化年龄。“第二代”表观遗传钟包括表观年龄和GrimAge。在513个CpG位点上训练PhenoAge并使用两阶段程序开发的报告。此外,PhenoAge在预测许多与年龄相关的疾病和寿命方面超过了“第一代”时钟的报告。GrimAge综合了1030个与吸烟包年相关的CpG和7种血浆蛋白的数据。尽管样本、方法和特征各不相同,但所有表观遗传钟均已验证了其准确评估表观遗传年龄的能力的报告。
我们认识到了现有研究在神经退行性疾病与EAA和寿命遗传相关性方面的一些局限性。首先,研究表明,表观遗传钟参与了一些神经退行性疾病,如AD、PD和肌萎缩侧索硬化症(ALS)。然而,据我们所知,目前还没有研究揭示MS和LBD与EAA和长寿的遗传关联。未被确认的因果关系和它们之间的遗传重叠促使我们进一步探索。其次,观察性研究可能存在未控制混杂因素的风险(例如,慢性炎症和药物摄入; ,从而掩盖了真实的的结果。随机对照试验(RCT)是证明医学相关暴露与结局之间因果关系的金标准。然而,RCT总是昂贵且持续时间长。此外,也很难确保研究符合伦理,并可扩展至其他人群。孟德尔随机化(MR)分析在流行病学、系统生物学、药物基因组学等领域具有克服不可测量的混淆和低成本的优点,已被广泛应用于因果推断。
在本研究中,我们旨在检测多种神经退行性疾病(AD、PD、LBD、ALS和多发性硬化症[MS])与四种表观遗传时钟(GrimAge、PhenoAge、IEAA和HannumAge)以及神经退行性疾病和多变量长寿相关表型(父母寿命、健康寿命和异常长寿)之间的因果关系和遗传病因学重叠(图1)。为了实现这一目标,我们主要利用MR和条件/联合错误发现率(cond/conjFDR)方法,使用大规模全基因组关联研究(GWAS)数据集。此外,我们鉴定了包括神经退行性疾病、表观遗传老化和多变量长寿相关表型的共享分子表型的多效性遗传变体、基因和生物学途径。
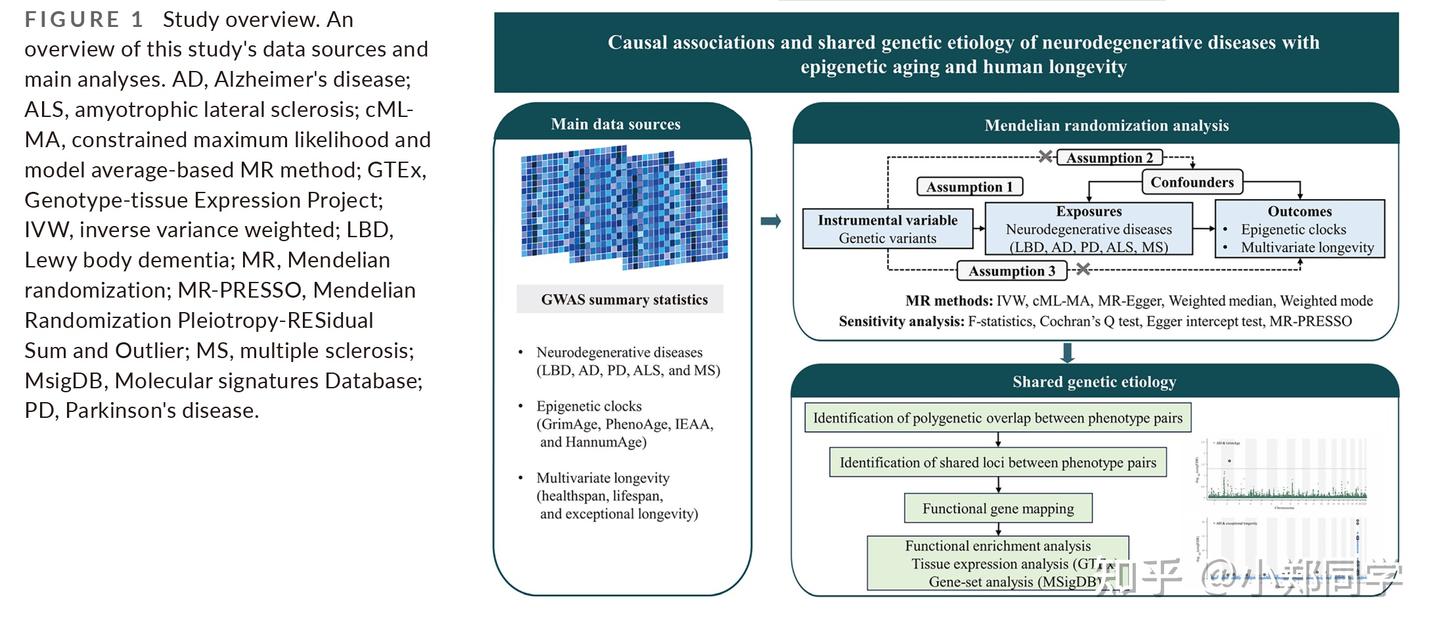
RESULTS
1、孟德尔随机化分析
我们鉴定了LBD的5个独立SNP、AD的33个SNP、PD的26个SNP、MS的94个SNP和ALS的7个SNP(表S1)。所有选择的独立SNP的F-SNP统计量均大于10(表S1)。没有SNP作为回文被丢弃。此外,这些SNP被用于MR分析。
显著证据显示,AD患者的异常寿命短0.309年(IVW β = −0.309,95% CI −0.38至−0.24,p = 1.51E-1919 < 3.33E-1903)(图2,表S2)。作为IVW方法的补充方法,基于约束最大似然和模型平均值的MR(cML-CRIMA)方法也显示了显著的因果效应(beta = −0.06,95% CI −0.07至−0.05,p = 3.31E-0.20 < 3.33E-0.03)(表S3)。这些p-ε值通过了多次校正测试。接下来,我们观察到AD和GrimAge年龄加速之间具有显著因果关系的证据(IVW beta = −0.10,95% CI −0.188至−0.013,p = 0.02; cML-ARMA beta = −0.04,95% CI −0.159至−0.0002,p = 0.04)(图2,表S3和S4)。敏感性分析的方向(加权中位数、简单模式和加权模式)与主要分析的估计效应相同。此外,我们没有发现其他神经退行性疾病对表观遗传衰老和人类寿命的任何强有力的因果影响(图S1和S2,表S2-S4)。

2、敏感性分析
为了确保上述提示性和显著性因果关系是稳健的,我们进一步进行了敏感性分析。我们进行的乘法随机效应IVW方法可以减少异质性引起的偏倚,以便更准确地估计因果效应。为了进一步检测是否存在异质性,Cochran's Q检验表明,在GrimAge年龄加速(p = 0.60)和异常寿命(p = 0.45)方面,AD的异质性不存在异质性(表S5)。我们没有发现AD和GrimAge年龄加速之间存在水平多效性(MR-PRESSO因果估计= −0.10,p = 0.69)。此外,根据 MR--PRESSO 分析,AD 和异常寿命之间没有水平多效性 (MR--PRESSO 因果估计 = ?0.31,p = 0.17) (表 S6)。总体而言,敏感性分析表明,AD对GrimAge年龄加速和AD对异常长寿的MR结果是稳健的。
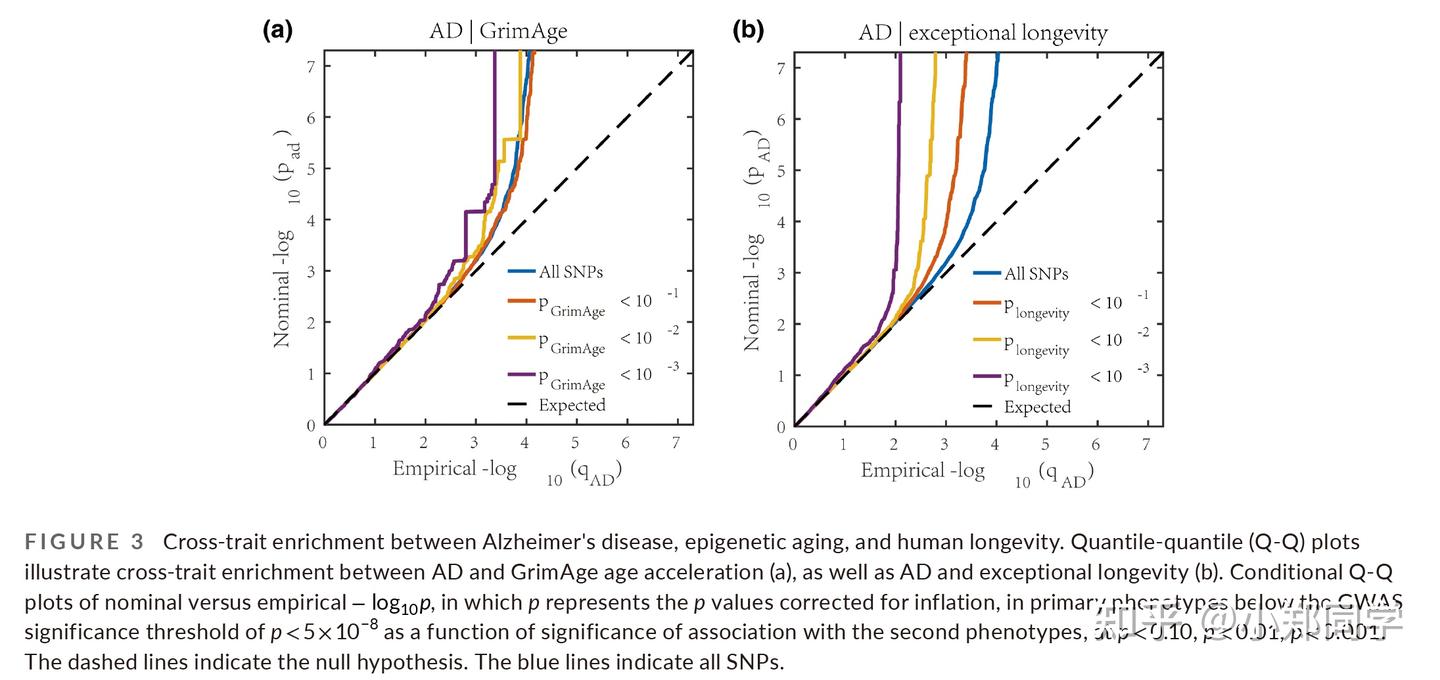
3、交叉性状富集
条件Q-IQ图说明了AD的SNP富集的连续增量作为与GrimAge(图3a)和异常长寿(图3b)的显著关联的函数,表明多基因重叠。反向条件Q-PQ图的富集结果与正向条件Q-PQ图的富集结果一致(图S3)。
4、阿尔茨海默病、表观遗传衰老和人类寿命之间的共享基因座
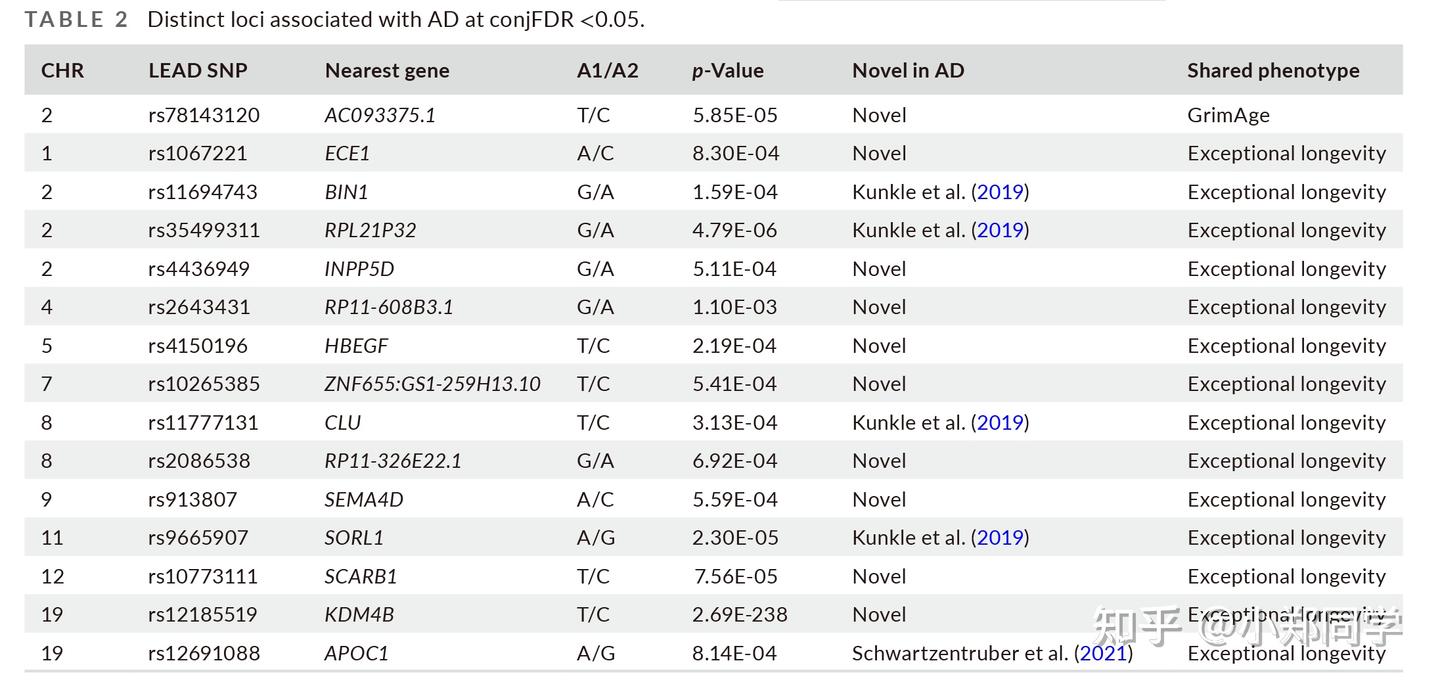
在确定多基因重叠的存在之后,我们进行双向交叉遗传性状富集以增加统计功效,并利用conjFDR分析来检测共享的基因组基因座。我们鉴定了一个不同的基因组位点(即,rs78143120)(图4a、表2、表S8)。共定位分析还表明rs78143120(PP.H4 = 0.82)是两个性状共有的基因座(表S9)。2号染色体上AC093375.1处的该位点是AD的新位点。此外,通过conjFDR鉴定了14个与AD和异常长寿相关的不同基因组基因座,其中9个被定义为新基因座(图4b,表2,表S10)。在这些基因座中,只有rs12691088(PP.H4 = 1)是通过共定位分析检测到的共有基因座(表S9)。
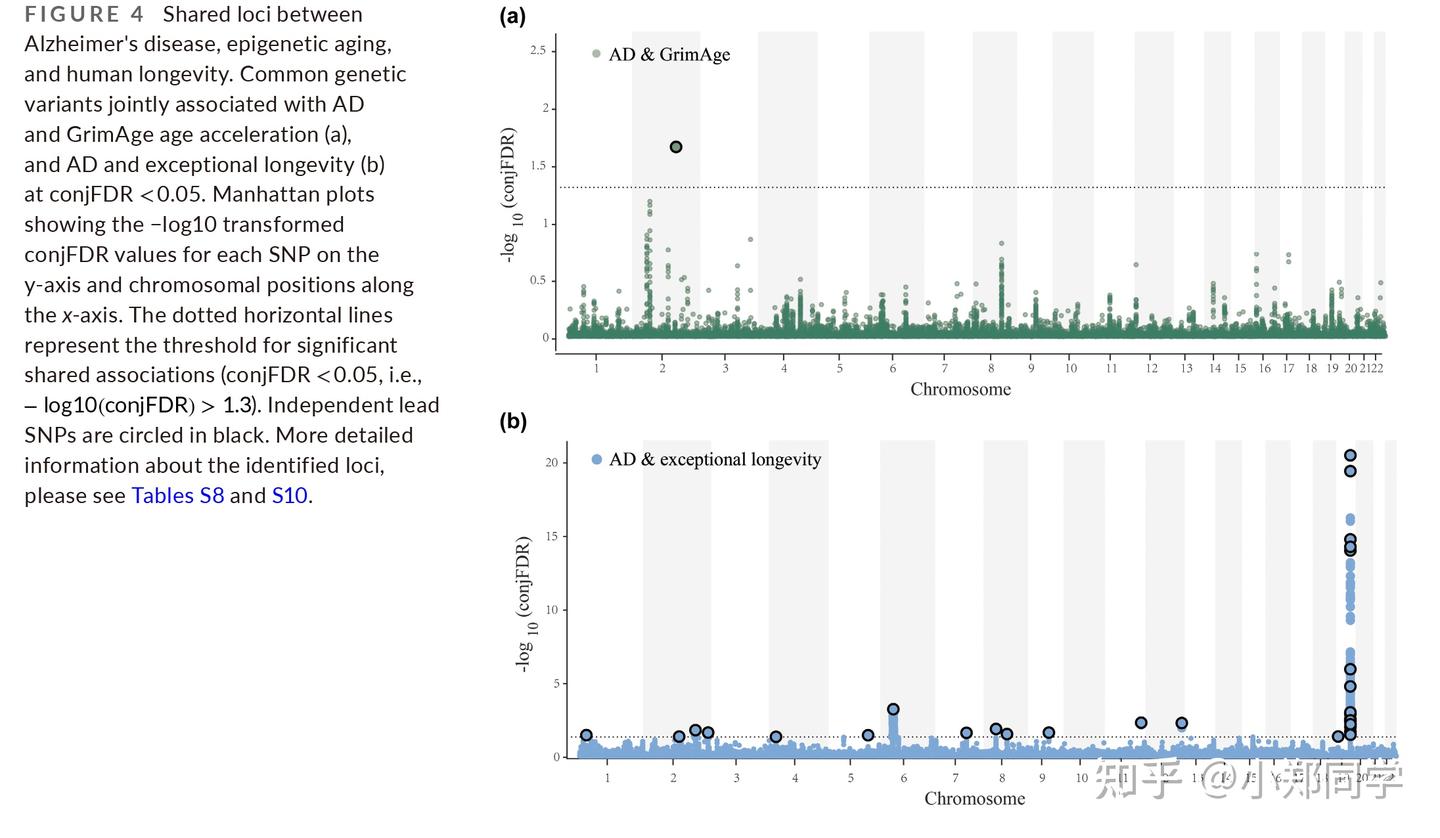
随后,我们评估了每个共享基因组基因座处的前导SNP的作用方向(表S8和S10)。14个共享的前导SNP中有8个在AD和异常长寿之间表现出一致的效应方向。此外,AD和GrimAge年龄加速之间共享的前导SNP的作用方向不一致。
5、功能注释
与AD和GrimAge年龄加速联合相关的候选SNP的功能注释表明,它们都位于基因间区域(表S11)。将与AD和异常长寿相关的10个候选SNP功能性注释到外显子区域(表S12)。在10个候选SNP中的7个中发现错义突变(表S13)。此外,AD和异常长寿之间共有的27个基因座具有CADD > 12.37,表明潜在的有害作用(表S12)。
此外,我们将7个和191个蛋白质编码基因分别定位到与AD和GrimAge年龄加速以及AD和异常长寿联合相关的候选SNP上(表S14和S15).一些基因定位于与AD和GrimAge年龄加速相关的基因座,以及AD和异常长寿与神经变性疾病相关,例如KCNJ 3和ERMN与癫痫相关; NR 4A 2参与伴有语言障碍的智力发育障碍和早发性多巴反应性肌张力障碍帕金森综合征; APOE,已知为AD的遗传风险因子的报告。
最后,我们鉴定了16个基因组,其显示出显著富集,其中基因定位于AD和异常长寿之间共有的基因座(表S16)。最重要的富集基因集是基因本体论 (GO) 生物过程的极低密度脂蛋白颗粒清除,以及 GO 细胞成分的蛋白-脂质复合物。由于定位的基因数量较少,因此无法对AD和GrimAge年龄加速之间的共享基因座定位的基因进行基因组分析。
DISCUSSION
据我们所知,这项研究是第一个调查神经退行性疾病和表观遗传年龄加速之间的因果关系和共同的遗传病因,以及神经退行性疾病和多变量寿命。有证据表明AD与GrimAge年龄加速和异常长寿有因果关系。在发现多基因重叠后,我们确定了AD和GrimAge年龄加速之间的一个共享基因组位点,以及AD和异常长寿的一个基因组位点。在这些不同的基因座鉴定,只有一个新的AD。有趣的是,我们发现一些显着丰富的基因组通过基因本体基因组分析的基因定位到AD和异常长寿之间的共享位点,这都与脂蛋白的合成和清除。
表观遗传钟在AD、PD、ALS等疾病中已得到应用,但在LBD、MS等疾病中的研究还不多见。一项横断面研究表明,表观遗传年龄与某些AD风险因素呈正相关,如BMI、总胆固醇与高密度脂蛋白胆固醇的比值等。McCartney 等人发现,11 名迟发性 AD 患者 (LOAD) 的表观遗传年龄比实际年龄年轻 20 岁。表观遗传加速的方向与我们的结果一致。然而,麦卡特尼等人研究的表观遗传年龄减速的年份与我们的研究大不相同。以下三个原因可以解释这一点:(1)表观遗传加速的程度在不同年龄组之间存在差异。McCartney等人的研究中LOAD样本的年龄约为90岁,比我们研究中的AD样本年龄大;(2)AD亚型(即,LOAD和AD)在表观遗传加速方面存在差异;(3)不同的表观遗传时钟可以预测不同的表观遗传年龄。McCartney等人在他们的研究中使用了Horvath时钟,而我们使用的是GrimAge,他们可能会捕捉到不同的衰老特征。此外,研究表明表观遗传时钟加速与PD发作时的年龄有关和ALS的发病年龄和预后。
菲利普等人已表明痴呆症与预期寿命减少有关。AD是最常见的痴呆症,占所有痴呆症病例的60%-80%。一项为期6年的跟踪研究表明,阿尔茨海默病患者在出现症状和确诊后的存活率都显著缩短。总体而言,我们的研究还发现,AD会减少预期寿命,这与之前的研究结果是一致的。
有些人可能会认为,AD对表观遗传年龄的四个指标的因果关系不一致,表明它们之间的因果关系是不确定的,不合理的。应当指出,这实际上并不矛盾。这是因为不同表观遗传时钟的计算模型基于不同的组织、DNA甲基化位点和训练以捕获不同特征的方法。
cond/conjFDR检测到的一些与AD和异常长寿相关的独特位点具有显著性,这与共定位分析的结果不一致。我们认为,不同方法之间发现的差异也可能意味着这些基因座与这两种性状都相关。例如,虽然仅通过cond/conjFDR方法鉴定为AD和异常长寿之间共有基因座的四个不同基因座(rs 11694743、rs35499311、rs 11777131和rs 9665907)未通过共定位分析检测到,但这四个基因座已显示与两种表型相关。因此,为了更稳健,我们仅认为通过两种方法鉴定的位点(rs78143120和rs 12691088)是性状之间共享的不同位点。对于其他13个位点,我们谨慎地假设它们可能是AD和异常长寿之间的共有位点,还需要更多的实验来证实。
在上述15个位点中,有10个位点被确定为AD的新位点。在这些新的基因座中,rs78143120定位于AC093375.1。一项跨性状Meta分析显示,rs61597598(AC093375.1)可能与打鼾和AD相关的报告。rs78143120是否与AD密切相关还需要进一步的实验证据来证实。rs 1067221定位于ECE 1(内皮素转化酶1),其可降解淀粉样蛋白β(Aβ)的报告。rs 4436949定位于INPP 5D,据报道其表达与AD风险相关,并由斑块相关小胶质细胞诱导。rs 4150196与智力显著相关的报告。众所周知,高智力和高认知能力降低了AD的风险,这支持了我们分析的可靠性。rs 10265385被定位于ZNF 655,其参与AD发病机理中的转录调节的报告。rs 913807基因定位于SEMA 4D,SEMA 4D是中枢神经系统疾病的关键因子。rs 9665907定位于SCARB 1(Aβ-受体清道夫受体B1)。SCARB-1蛋白表达减少会增加Aβ斑块沉积,但对Aβ斑块周围的小胶质细胞蓄积没有影响,实际上会加重学习和记忆中的认知缺陷。rs 12185519被定位于KDM 4 B,KDM 4 B具有通过阻断ICAM 1和VCAM 1-诱导的外渗来抑制脑部疾病(例如AD)的潜力。这些新的基因座都与神经疾病的途径或原因相关。需要进一步的实验和研究来证实。
本研究存在一些局限性。首先,没有个人层面的数据。因此,我们无法对不同年龄组的神经退行性疾病患者进行更详细的分析。其次,为了尽量减少由于人群分层而产生的偏倚,本研究仅纳入欧洲血统的个体。需要进一步的研究来证实我们在其他人群中的结果。第三,EAA或减速可能发生在患有神经退行性疾病的参与者中,这可能会使遗传重叠的结果产生偏差。然而,这种潜在的偏见不能解释共享位点之间的混合模式的影响方向。最后,鉴于我们发现的一些新基因座的遗传证据不足,我们认为这一结果应谨慎解释,需要更多的实验研究来验证。
总之,本研究证明了神经退行性疾病与表观遗传衰老和人类寿命的因果关系和共同的遗传病因。我们确定了两个具有混合效应方向的共同风险位点,其中一个被定义为AD的新风险位点。然而,需要进一步的生物学证据来揭示潜在的机制。
MATERIALS AND METHODS
1、神经退行性疾病GWAS数据集
我们获得了5种神经退行性疾病(LBD、AD、PD、ALS和MS)的公开可用GWAS汇总统计量。与LBD相关的SNP的汇总统计来自已发表的全基因组分析,其中包括2591例临床诊断为LBD的病例和4027例对照。我们从国际阿尔茨海默病基因组学项目(IGAP)、国际帕金森病基因组学联盟(IPDGC)、ALS变体服务器(AVS)和国际多发性硬化症遗传学联盟(IMSGC)(国际多发性硬化症遗传学,2019)分别(表1)。有关每个数据集的样本量和更多信息,请参见表1。上述数据集中的所有参与者主要是欧洲血统。
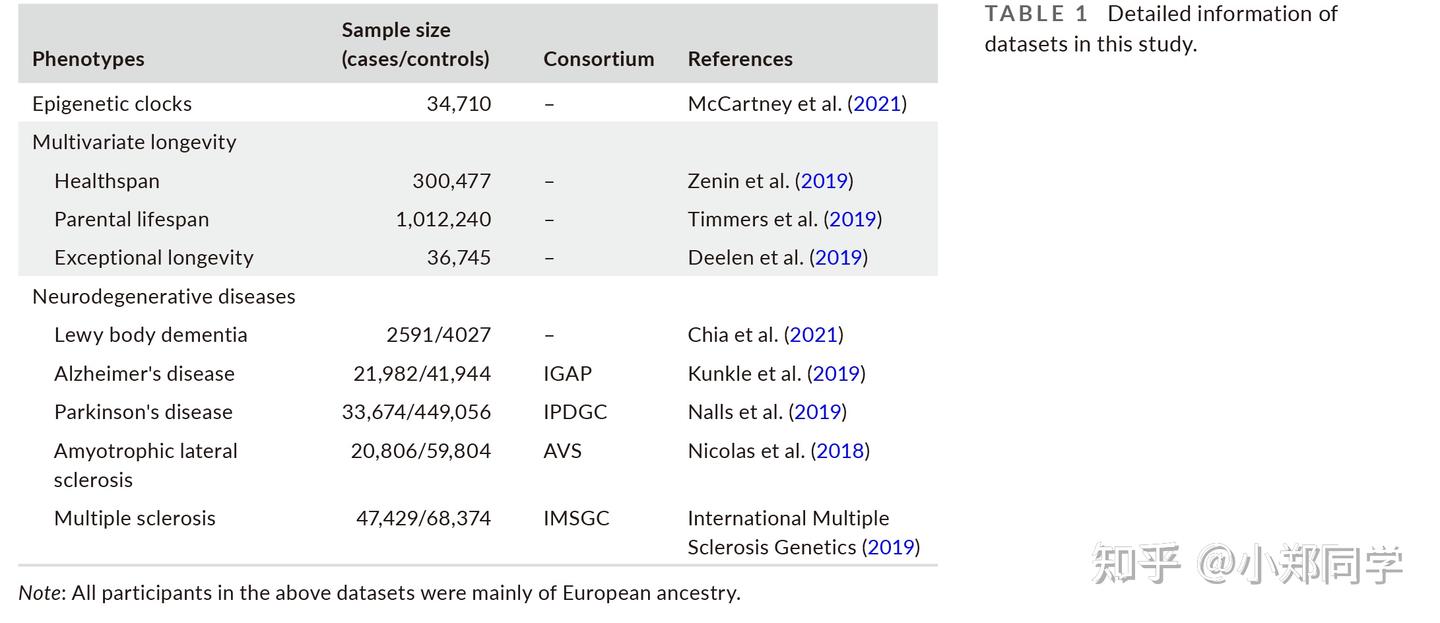
2、表观遗传时钟GWAS数据集
我们获得了表观遗传钟的四个汇总水平数据集(汉纳姆年龄、IEAA、表型年龄和格里姆年龄)(表1)。每个数据集包含34,710名欧洲血统的参与者。简而言之,McCartney等人使用Horvath表观遗传年龄计算器软件(一个在线工具),评估并计算了我们之前提到的四个表观遗传时钟的基于年龄调整的DNA甲基化指标。遗传学。加州大学洛杉矶分校edu/)或来自Steve Horvath和Ake Lu的独立脚本。随后,进行离群值样本过滤、质量控制和插补基因型。使用单倍型参考联盟或1000个基因组3期组进行基因型插补。性别和遗传主成分的线性模型进行调整。有关研究设计和实验程序的更多详细信息,请参阅McCartney et al.。
3、多变量寿命GWAS数据集
为了探索衰老的更临床相关的表型并进一步分析这些表型与神经退行性疾病之间的遗传关联,我们从与人类寿命相关的性状获得了多变量GWAS汇总统计量(N总= 1,349,432,N有效= 709,709)。多变量GWAS汇总-分级数据集包括四种老化表型,其是healthspan、亲代寿命和极长寿命(表1)。数据集中的所有参与者都是欧洲血统。
4、工具变量选择
神经退行性疾病的遗传工具变量通过全基因组显著性(p < 5.00E-08)和连锁不平衡(LD)聚集临界值(r2 < 0.01)来鉴定。通过对SNP数量的Bonferroni校正,调整了全基因组显著性的阈值(p < 5.00E-08)。它已成为GWAS的标准显著性阈值,尤其是对于欧洲人群的报告。删除回文SNP以避免偏倚。F-σ统计量是MR分析中仪器强度的一种度量。F > 10 表示该工具具有足够的强度来可靠地估计因果效应。我们通过公式(1)(β,效应量; SE,标准误)计算了F-统计量,并认为F-统计量大于10的工具变量与暴露量密切相关(即,神经变性疾病)(Burgess等,2011年)的报告。
5、孟德尔随机化分析
MR分析是一种流行病学方法,利用遗传变异为暴露(一种风险因素)和结局变量之间的因果关系提供统计学证据的报告。MR分析需要以下三个假设,以确保其结果稳健。第一,遗传变异体(即,工具变量)应当与暴露显著相关(例如,神经退行性疾病),其基因组范围内的显著水平通常为5.00E-08。其次,工具变量应独立于暴露的混杂因素(神经退行性疾病)。第三,工具变量应仅对暴露的结果变量(表观遗传钟和多变量寿命)产生影响。后两个假设统称为与多效性无关。
主要的MR分析方法是乘性随机效应逆方差加权(IVW)MR。IVW MR通过工具变量结合了神经退行性疾病对EAA和多变量寿命影响的遗传预测的报告。对主要IVW结果的多重检验应用Bonferroni校正。多重检验校正的显著性阈值计算为p = 0.05/(Nexposure × Noutcome),其中Nexposure和Noutcome分别是暴露变量和结局变量的数量。Nexposure × Noutcome是执行的测试次数。因此,Bonferroni-校正后的表观遗传钟和多变量寿命的显著性阈值分别为p = 0.05 5×4 = 2.50E-03和p = 0.05 5×3 ≈ 3.33E-03。主要MR分析由“TwoSampleMR”R软件包完成。此外,我们使用R软件包“Meta”进行固定效应Meta分析,汇总了神经退行性疾病的MR结果。
我们还使用MRcML R软件包进行了基于约束最大似然和模型平均值的MR方法(MR-cML-MA)作为二次MR分析,以控制相关和不相关的多效性效应。
6、敏感性分析
我们进行了MR-Egger、加权中位数和加权模式方法作为敏感性分析,以确定IVW MR效应估计的可靠性。使用“MR-PRESSO”R程序包。
7、错误发现率和共定位分析
我们进行了condFDR和conjFDR方法以改进与具有强因果关系的表型联合相关的特定基因组基因座的发现的报告。CondFDR统计框架建立在标准FDR的基础上,并纳入了主要目标性状的遗传汇总统计(例如:LBD)和条件性状(例如,GrimAge年龄加速)来重新调整初级表型中的检验统计量。condFDR < 0.01的遗传变异被认为与原发表型相关。ConjFDR是condFDR的一个进化,它应用两个性状之间的交叉性状富集来增加遗传发现。conjFDR < 0.05的遗传变异被认为是共享基因座的报告。随后,基于1000基因组计划LD结构,将识别出的显著遗传变异聚类到LD r2 > 0.1的LD区组中。此外,我们还进行了条件分位数-分位数(Q-Q)作图,以估计多效性富集和可视化交叉性状富集。Q-Q图说明了主要表型的p-值分布(例如,LBD),以及通过它们与次要表型。考虑到MHC区域的复杂和不寻常的LD模式和遗传结构,在拟合condFDR和conjFDR模型之前,我们基于人类基因组19个位置排除了扩展的主要组织相容性复合体(MHC)区域(chr 6:25119106-33854733)、染色体8p23.1(chr 8:7200000-12500000)和MAPT区域(chr 17:4000000 -47000000)周围的SNP,以避免偏倚。
我们还采用共定位分析来检测通过cond/conjFDR方法鉴定的独特的共有基因座是否对两个性状都起作用的报告。基于贝叶斯算法,共局部化方法计算了五个假设的后验概率。如果H4的后验概率> 0.75,则认为一个位点与两个性状都相关。共定位分析由R包“coloc”使用默认参数进行。
8、基因座定义
独立基因组基因座的定义与FUMA一致方案。独立的显著SNP被认为是conjFDR < 0.05和r2 < 0.6。此外,将这些SNP中r2 < 0.1的SNP确定为前导SNP。通过LD r2 ≥ 0.6识别候选SNP,其中具有独立的显着SNP。如果基因座之间的距离< 250 kb(即 LD 块相距 < 250 kb),则它们被合并。选择具有最显著conjFDR的SNP作为合并基因座的前导SNP。通过在r2 ≥ 0.6处鉴别LD中的所有候选SNPs与该基因座中的一个独立显著SNPs来定义基因组基因座的边界。LD信息由1000基因组计划参考小组的报告。表型间共有基因座的方向效应通过比较它们的z-评分和优势比来评估。如果新基因座与原始GWAS(±500 kb)中报告的基因座在物理上不重叠,或在NHGRI-EBI目录中未报告,则定义为新基因座。
9、功能注释
我们使用组合的注释依赖性缺失评分(CADD)注释SNP regulomeDB评分和染色质状态。CADD是一种流行的工具,它使用机器学习方法来计算每个基因组位点的CADD得分,以评估致病性的程度。RegulomeDB评分是基于RegulomeDB的评分,用于评估每个非编码变体的潜在功能。我们还利用FUMA和基因型组织表达(GTEx)资源来实现基因组富集并评估表达数量性状基因座(eQTL)功能。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号