
调节铁凋亡的分子生态系统研究进展
同学们,大家好!今天给大家介绍一篇综述,调节铁凋亡的分子生态系统研究进展,想了解铁死亡及其分子机制的同学们可以重点关注一下。这篇文章是2024年9月份发表于NATURE CELL BIoLogy(IF:17.3,中科院分区:生物学-细胞生物学 1区),下面我们详细看看。

在该综述中,作者分以下几个方面对铁死亡及相关分析机制进行了阐述:
1、铁死亡的核心机制,以及铁死亡启动的三个基本因素(活性氧、可氧化脂质、脂质过氧化);
2、铁凋亡中的抗氧化系统:酶抗氧化剂、非酶抗氧化剂、金属螯合剂、转录调节子;
3、膜修复系统;
4、治疗机会及挑战:机会(在肿瘤学、神经退行性疾病和I/R损伤中的应用)、挑战(特异性和选择性、药物递送、生物标志物鉴定、副作用、临床转化)
5、结论与展望
以下是文章具体内容:
A guideline on the molecular ecosystem regulating ferroptosis
Abstract
铁凋亡是一种以不受控制的脂质过氧化为特征的复杂调控的细胞死亡形式,自2012年以来,这个术语已经引起了人们的极大兴趣。近年来,在阐明铁凋亡诱导和防御的详细分子机制方面取得了显着进展,特别强调异质性和可塑性的作用。在铁凋亡的分子生态系统中,目前和未来的进展有望在广泛的疾病中解锁安全有效的治疗策略。
Introduction
铁凋亡(Ferroptosis)是在癌细胞中观察到的一种不同形式的调节性细胞死亡,依赖于铁,但与凋亡和坏死性凋亡不同。与依赖于成孔蛋白的溶解性细胞死亡机制不同,铁凋亡是由有毒的氧化脂质及其副产物驱动的,特别是4-羟基壬烯醛(4 HNE),沿着通过共价结合亲电脂质过氧化分解产物形成的脂化蛋白。亚铁凋亡在疾病的临床前研究中具有重要意义,包括癌症、神经退行性疾病和与缺血-再灌注(I/R)损伤相关的病症。它提供了一种治疗方法,用于治疗缺乏抗肿瘤的耐药癌细胞,而其抑制具有管理感染相关疾病,与铁过载或脂质毒性相关的无菌炎症的潜力。此外,铁凋亡在组织稳态和发育中起着至关重要的作用。在这篇综述中,我们的目的是提供一个更新的概述,涵盖其基本机制,异质性和可塑性。我们也将深入研究整合的抗氧化剂和膜系统在调节铁蛋白敏感性中的作用,沿着讨论疾病的影响、治疗前景和相关的挑战。铁凋亡的核心机制
Erastin和RSL3是用于诱导铁凋亡的常见小分子。这些化合物最初是在针对RAS突变癌细胞的筛选中发现的,它们触发了一种非凋亡的铁依赖性细胞死亡形式,导致了术语“铁凋亡”。与此同时,GPX4的遗传失活被发现可诱导氧化性非凋亡性细胞死亡和系统xc −的过表达,以保护细胞免受类似的非凋亡性细胞死亡,突出了该过程作为靶向RAS突变的潜在癌症治疗的可能性,同时保留正常细胞。
进一步的研究表明,铁凋亡是高度依赖于环境,金属离子,如锌和铜,除了铁,可以在特定条件下诱导铁凋亡。RAS野生型和突变型细胞,包括癌细胞和非癌细胞,都可以发生铁凋亡。在各种器官(例如,肾)或细胞(例如,T细胞或B细胞)可引起铁毒性损伤,突出了其在发育生物学中的作用。
铁凋亡与自噬密切相关,自噬水平升高通常与铁凋亡敏感性增加相关。特定类型的选择性自噬,如铁蛋白吞噬,脂肪吞噬和时钟吞噬,可导致铁积累和脂质过氧化,诱导铁凋亡。人类神经元中的全基因组CRISPRi/a筛选揭示了所谓的ATG(自噬相关)家族成员(例如,BECN 1 [beclin 1])和溶酶体蛋白(例如,PSAP [saposin原])通过触发脂褐素的形成或增加铁积累参与铁凋亡。在某些情况下,ATG基因的缺失对细胞死亡没有影响,包括铁凋亡。这些发现强调了铁凋亡的适应性和环境依赖性,但它的启动涉及三个基本要素,这将在下面讨论。活性氧
铁凋亡诱导中的第一个关键因素是存在刺激各种来源的ROS产生的起始信号(图1):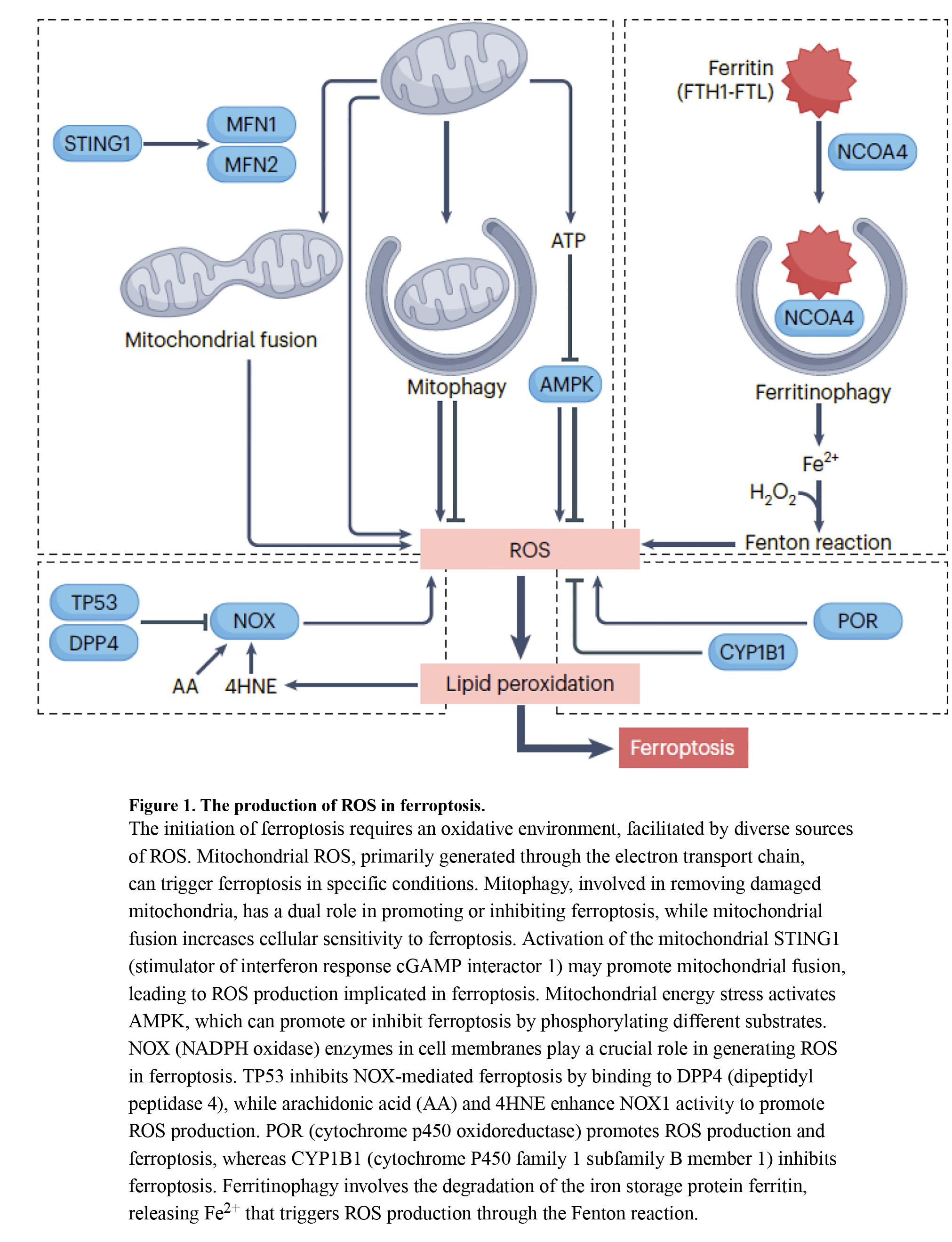
1)线粒体:线粒体是 ROS 的主要来源,主要是氧化磷酸化过程中的超氧阴离子/O2。线粒体 SOD 将超氧化物转化为其他 ROS,包括过氧化氢 (H2O2)。线粒体ROS可以触发铁凋亡,其中精氨酸分解促进由脱胱氨酸引起的铁凋亡。线粒体质量受线粒体自噬调节,线粒体自噬在铁凋亡中具有双重作用。线粒体分裂促进细胞凋亡,而线粒体融合可增加细胞对铁中毒的敏感性。线粒体能量应激通过AMPK介导的ACACA/ACC(乙酰辅酶A羧化酶α)磷酸化抑制铁细胞凋亡,但AMPK也可通过靶向BECN 127或通过破坏嘧啶体组装,阻碍嘧啶中间体合成,促进铁细胞凋亡。
2)NOX(NADPH氧化酶):NOX的过表达增加ROS水平,提高铁凋亡敏感性。NOX在铁凋亡中的活性受多种因素调节,如TP 53(肿瘤蛋白p53)和ALDH1B1(醛脱氢酶1家族成员B1)。Trp 53/TP 53缺乏促进DPP 4(二肽基肽酶4)在细胞膜上的积累,与NOX1形成复合物,并导致亚铁蛋白变性死亡。ALDH1B1通过催化醛的氧化,将其转化为羧酸,抑制NOX1活性的铁中毒诱导作用。
3)酶促反应:ROS可以是酶促反应的副产物,如参与药物代谢的细胞色素P450及其还原酶。POR(细胞色素P450氧化还原酶),一种黄素蛋白,通过产生超氧化物自由基诱导脂质过氧化和铁凋亡。
4)芬顿反应。该反应涉及H2O2与过渡金属(通常为铁(Fe 2+))之间的相互作用,导致产生高反应性羟基自由基/·OH。在铁凋亡期间广泛研究的铁代谢机制是铁蛋白自噬,其中自噬降解铁储存蛋白铁蛋白。这释放游离铁,将一种ROS类型转化为另一种,从而在癌细胞和非癌细胞中诱导铁凋亡。
可氧化脂质
铁凋亡的第二个关键因素是易氧化的多不饱和脂质的存在(图2)。细胞膜是铁凋亡中氧化损伤的主要靶点,可受到促进脂质合成的代谢途径的影响,特别是多不饱和脂肪酸(PUFA)的产生,增加细胞对铁凋亡诱导剂的敏感性。虽然引发铁凋亡所需的PUFA分解的确切阈值仍然不清楚,但一个公认的正调节因子是ACSL4。ACSL4通过将长链脂肪酸转化为酰基辅酶A酯来激活长链脂肪酸,促进其进入各种代谢途径。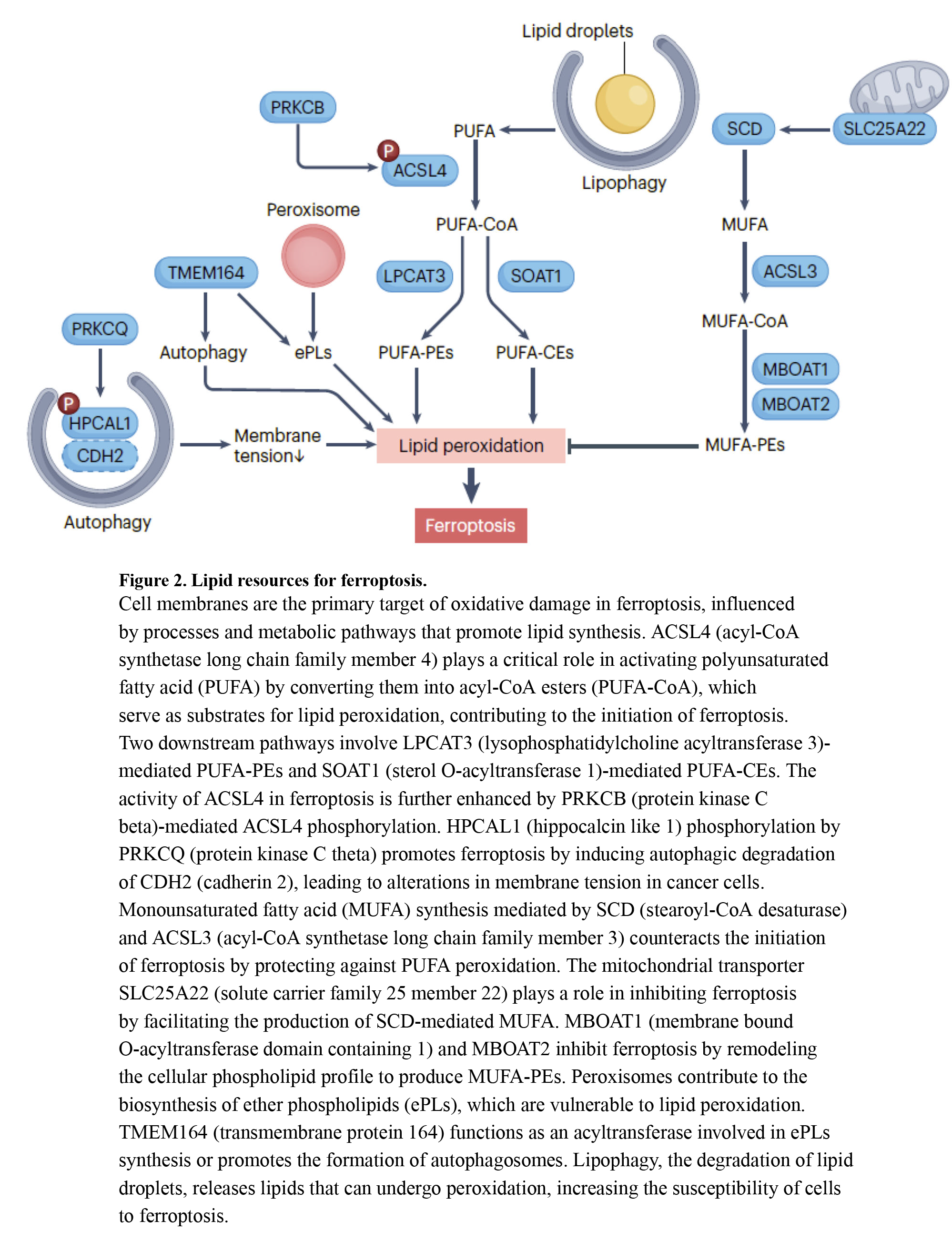
ACSL4介导两个下游途径,产生不同的PUFA相关酰基辅酶A酯。一个涉及LPCAT3(溶血磷脂酰胆碱酰基转移酶3),将PUFA掺入磷脂酰乙醇胺(PE,而另一个激活SOAT1(甾醇O-酰基转移酶1),产生PUFA-胆固醇酯(CE)而不是PUFAPE。这两种途径都有助于脂质过氧化,根据上下文作为底物。在脂质翻转酶SLC47A1缺陷的人胰腺癌细胞中,ACSL4驱动的PUFA-CE产生是特别相关的。ACSL4 激活是一种通过诱导实体癌铁死亡来提高化疗或免疫治疗疗效的策略。PRKCB/PKCβII通过Thr 328磷酸化增强ACSL4活性,而PRKCQ在Thr 149处的HPCAL 1磷酸化通过CDH2的自噬降解诱导铁凋亡,从而改变癌细胞中的膜张力。
ACSL3合成单不饱和脂肪酸(MUFA),可竞争性抑制PUFA过氧化,防止铁凋亡启动。线粒体谷氨酸转运蛋白SLC 25A22通过增强GSH和MUFA合成来抑制胰腺癌细胞中的铁凋亡。性激素受体上调的MBOAT1(膜结合O-酰基转移酶结构域1)和MBOAT2通过重塑细胞磷脂谱以产生含MUFA的磷脂来抑制癌细胞中的铁凋亡。ACSL4-独立途径增加了对细胞死亡调节中脂质代谢的理解的复杂性。
过氧化物酶体,参与脂肪酸分解,过氧化氢的产生,和PUFA缩醛生物合成,可以增加铁凋亡的敏感性。它们还含有抗氧化酶,如CAT,可抑制铁凋亡,以及MUFA缩醛磷脂,可预防铁凋亡。因此,过氧化物酶体或缩醛磷脂会根据上下文积极或消极地影响铁凋亡。
脂质吞噬选择性降解脂滴,释放脂质进行过氧化,使细胞,特别是肝癌细胞,更容易发生铁中毒。ACSL3增加脂滴中的脂质储存可以限制透明细胞肾细胞癌细胞中的铁凋亡。
此外,TMEM164通过充当酰基转移酶、合成C20:4醚磷脂和促进膜驱动的吞噬细胞的形成而充当铁凋亡的正调节剂。这些吞噬细胞对于胰腺癌细胞响应于铁凋亡刺激而不是营养饥饿随后产生自噬体是必不可少的。
脂质过氧化
几种酶,包括ALOX、PTGS/环氧合酶和细胞色素P450酶,在铁凋亡期间催化脂质过氧化中起关键作用(图3)。
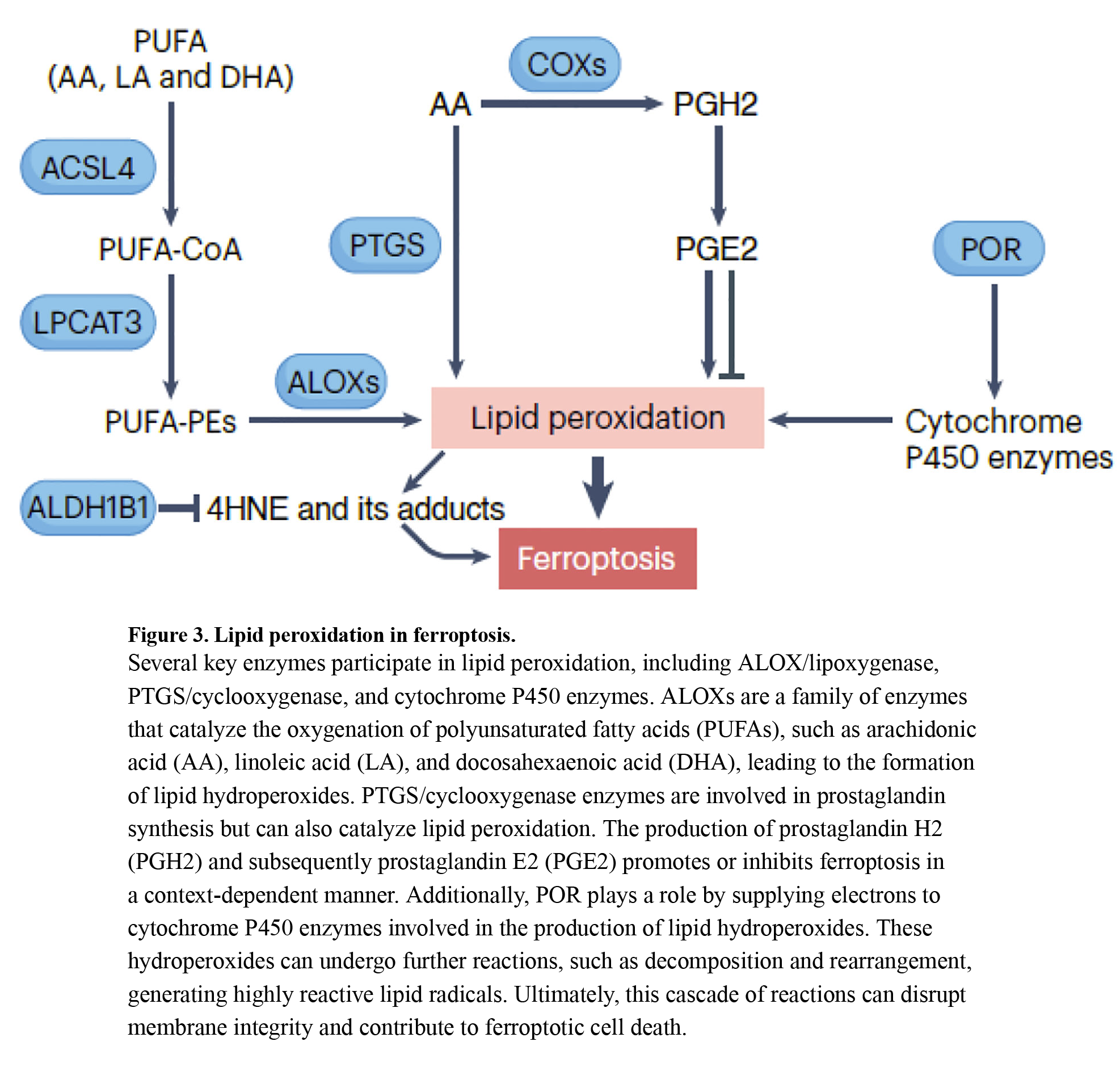
ALOX是催化PUFA氧化的酶,通过将氢过氧基(-OOH)引入脂肪酸链来引发脂质过氧化。人类有六种ALOX同种型(ALOX 5、ALOX 12、ALOX 12 B、ALOX 15、ALOX 15 B和ALOX 3),具有不同的底物偏好和催化活性,导致各种细胞或组织中的铁凋亡。PEBP1(磷脂酰乙醇胺结合蛋白1)与ALOX15形成催化复合物,有效地过氧化PUFA-PE。靶向ALOX15-PEBP1复合物的抑制剂可有效防止磷脂过氧化,并减轻体内全身照射造成的损伤。然而,Alox15的缺失并不能阻止肾脏或T细胞中Gpx4缺失驱动的铁凋亡。因此,分析实验模型中的ALOX表达对于评估铁凋亡中不同ALOX成员的需求至关重要。
PTGS/环氧合酶通过氧化游离PUFA催化脂质过氧化,产生脂质氢过氧化物。然而,它们的主要功能是合成前列腺素,在脂质过氧化中起次要作用。PGE 2的产生可以通过PTGER 1和PTGER 2抑制脑I/R54中的铁凋亡,但在急性肾损伤中促进铁凋亡。
参与药物代谢的细胞色素P450酶可以通过将氧引入脂肪酸链中来催化脂质过氧化,产生脂质氢过氧化物和4HNE(已知的铁凋亡介体)。如前所述,POR通过向分子氧提供电子,促进H2O2产生以诱导铁凋亡而发挥作用。
不管催化脂质过氧化的酶是什么,脂质氢过氧化物都会引发连锁反应。它们经历裂解反应,通常由铁等过渡金属催化,产生高度反应性的脂质自由基。这些自由基与附近的脂质反应,在自蔓延过程中放大脂质过氧化作用。然后形成亲电的、氧化性截短的磷脂变体,与蛋白质中的氨基酸残基反应以诱导蛋白质脂质氧化。这一系列反应破坏细胞膜,改变膜张力,损害膜修复,并最终导致铁中毒质膜透化。ER被认为是可能导致其他细胞器随后氧化膜损伤的初始位点。
铁凋亡中的抗氧化系统
酶抗氧化剂
参与抗氧化防御铁凋亡的关键酶是GPX 4,它将生物膜中的脂质氢过氧化物还原为醇(图4)。GPX 4的活性中心含有硒代半胱氨酸。低硒水平导致核糖体在GPX 4的无效解码硒代半胱氨酸UGA密码子处停滞,引起核糖体碰撞、过早翻译终止和N-末端GPX 4片段的蛋白酶体清除。分子伴侣HSPA 5直接稳定GPX 4蛋白,而自噬或泛素-蛋白酶体系统介导GPX 4蛋白降解,增加铁凋亡敏感性。CKB介导的GPX 4丝氨酸残基的磷酸化抑制自噬介导的GPX 4降解和随后的铁凋亡。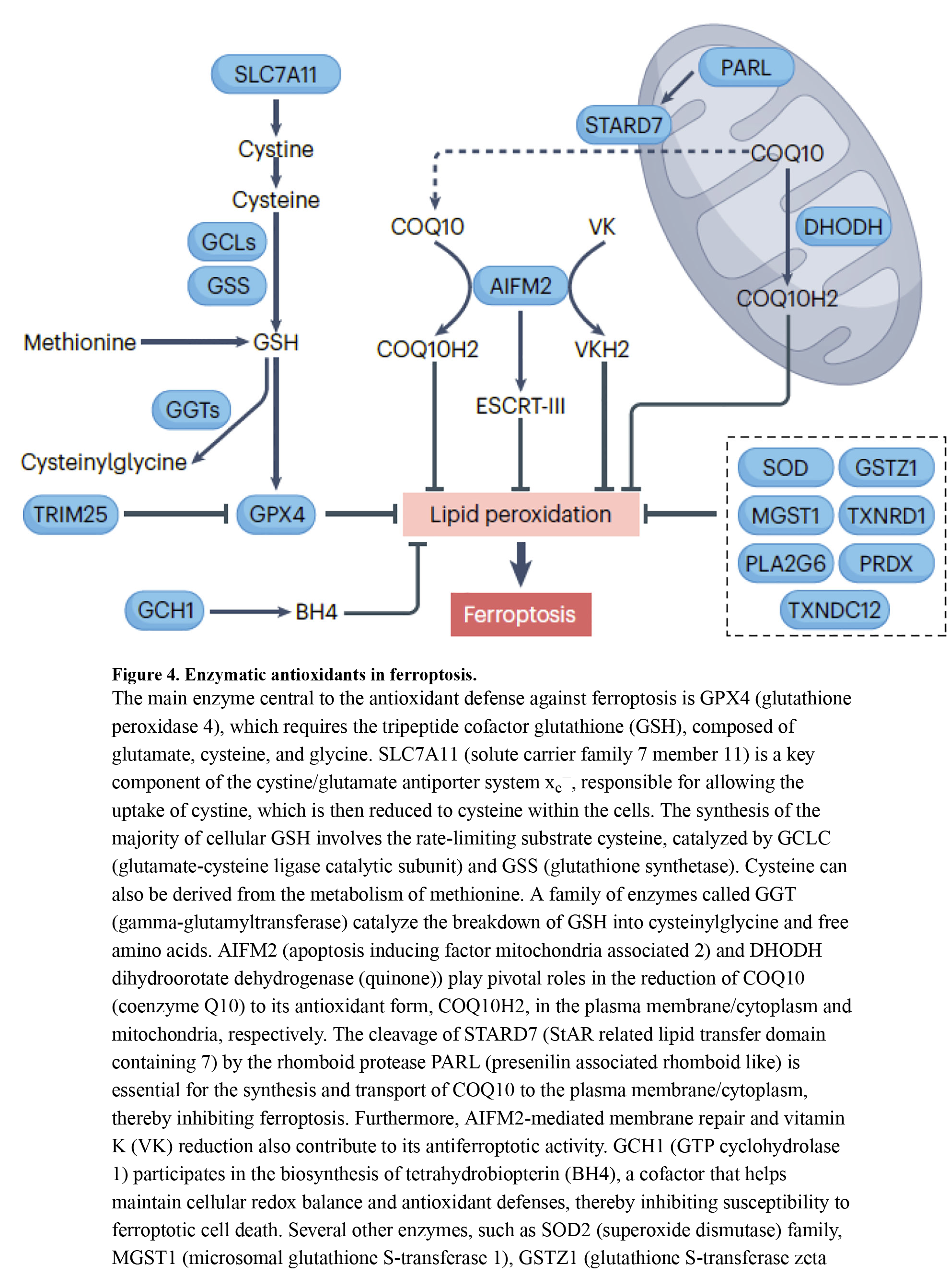
GPX 4中的R152 H突变可导致Sedaghatian型脊柱干骺端发育不良/ SSMD,这是一种新生儿罕见且致命的疾病。体外研究表明,这种R152 H突变不会直接影响酶的催化活性,而是干扰心磷脂对其变构激活。进一步的检查是必要的,以确定是否过度的心磷脂过氧化功能障碍的线粒体GPX 4有助于疾病的发展。
组成型敲除小鼠中的Gpx 4基因导致约7.5-8.5天的胚胎死亡。体内证据表明,Gpx 4缺乏症与铁凋亡有关,这首先是在肾脏中Gpx 4条件性敲除的小鼠中观察到的,结合维生素E缺乏饮食,导致肾脏损伤。这种表型被维生素E补充剂或铁凋亡抑制剂livestatin-19逆转。类似地,在缺乏Gpx 4的情况下,小鼠中活化T细胞的铁凋亡可通过富含维生素E的饮食来预防。在正常的繁殖条件和食物喂养下,几种细胞类型(例如骨髓、胰腺上皮细胞或肝细胞)中 Gpx4 的条件敲除不是致命的。然而,在标准食物饮食下,神经元中Gpx4的可诱导条件性敲除或肠上皮中Gpx4的纯合条件性缺失是致死的。因此,GPX4及其对脂质过氧化的防御在调节组织发育中起着依赖于环境的作用。
GSH是由谷氨酸、半胱氨酸和甘氨酸组成的三肽,作为GPX 4辅因子发挥作用。半胱氨酸是GSH合成的关键前体,可限制GSH的产生,并来源于蛋氨酸代谢。此外,更重要的是,细胞通过胱氨酸/谷氨酸反向转运体系统xc−(由SLC 7A 11和SLC 3A 2亚基组成)输入细胞外胱氨酸。输入的胱氨酸随后被还原为半胱氨酸。药理学药物如erastin或柳氮磺胺吡啶可以抑制系统xc−。据报道,在高浓度下,索拉非尼以间接方式抑制系统xc −的活性,但最近的一项研究表明,索拉非尼仅不能诱导某些癌细胞中的铁凋亡。GSH主要通过酶促反应在胞质溶胶中合成,系统xc−对于维持GSH水平以防止铁凋亡在其开始之前至关重要,因为铁凋亡发作期间GSH合成太慢。
尽管GSH耗竭导致铁凋亡,但GPX 4不是GSH的唯一靶点,这表明存在GPX 4非依赖性保护途径来对抗铁凋亡(图4)。其中,AIFM 2/FSP 1在Gpx 4缺陷细胞中从线粒体重新定位到细胞膜,减少COQ并抑制铁凋亡。STARD 7(含有7的星星相关脂质转移结构域),在PARL(早老蛋白相关菱形样)切割后在线粒体膜间隙和细胞溶质中发现,参与COQ 合成和转运到质膜,也阻碍铁凋亡。此外,AIFM 2有助于膜修复和经典维生素K循环,增强其抗铁凋亡作用。AIFM 2在铁凋亡中的活性依赖于相分离,并且可以通过化合物icFSP 186促进的N-末端肉豆蔻酰化来引发。
DHODH(二氢乳清酸脱氢酶(醌))是一种参与嘧啶生物合成的线粒体酶,对DNA和RNA的形成至关重要。DHODH的活性对表达低水平GPX 4的癌细胞的铁敏感性有影响,这可能是由于DHODH催化利用COQ作为电子受体。抑制DHODH会降低辅酶Q,增加对脂质过氧化和铁凋亡的易感性。然而,DHODH抑制剂对AIFM2的潜在脱靶效应仍有争议。
GPX 4、AIFM 2和DHODH之外的几种抗氧化酶在抑制铁凋亡中发挥作用。GCH 1(GTP环化水解酶1)参与四氢生物蝶呤/BH 4生物合成,有助于细胞氧化还原平衡和铁凋亡抑制。线粒体SOD 2防御热应激诱导的铁凋亡。NOS 2/iNOS(一氧化氮合酶2)通过抑制ALOX 15介导的脂质过氧化来抑制巨噬细胞中的铁凋亡。NFE 2L 2/NRF 2介导的MGST 1上调有助于胰腺癌细胞对促铁激活剂的细胞解毒。GSTZ1/马利乙酰乙酸异构酶(谷胱甘肽 S-转移酶 zeta 1)抑制膀胱癌细胞的铁死亡,而 TXNRD1(硫氧还蛋白还原酶 1)、TXNDC12(硫氧还蛋白结构域包含 12)和过氧化物酶 (PRDX) 在铁死亡抑制中也具有上下文依赖性作用。此外,Ca 2+非依赖性PLA 2G 6/iPLA 2 β/PNPLA 9(磷脂酶A2 VI组)通过水解过氧化膜磷脂(可能由TP 53调节介导)在消除铁中毒死亡信号中发挥作用。理解不同抗氧化系统在铁凋亡中的协同作用仍然是转化医学的中心主题或挑战。
非酶抗氧化剂
非酶抗氧化剂抵消有害的ROS并保护细胞免受氧化损伤,维持细胞氧化还原平衡。铁缺乏症的例子包括维生素E、维生素K、GSH、COQ和NADPH。它们与酶抗氧化剂合作,以防止或减轻氧化应激。抗氧化剂在还原时会抑制自由基,但其氧化形式可能会增加氧化应激,强调动态监测氧化还原反应的重要性。金属螯合剂
铁和铜等金属离子参与芬顿或哈伯-韦斯反应,产生高活性羟基自由基。金属结合蛋白,如TF(转铁蛋白)和铁蛋白,螯合游离铁,以防止这些破坏性反应。细胞内金属稳态由专门的蛋白质严格调节,包括将金属递送至其靶蛋白的金属分子伴侣。金属硫蛋白还有助于控制金属离子的可用性,减少其对氧化损伤和铁中毒的贡献。此外,金属螯合剂药物如去铁胺、去铁酮、地拉罗司和环吡酮,在临床环境中使用,已经显示出通过对抗脂质过氧化过程来调节铁凋亡的前景。转录调节子
NFE 2L 2:在氧化应激或暴露于亲电子化合物时,NFE 2L 2从KEAP 1释放并易位到细胞核中。SQSTM 1(隔离体1)介导的蛋白质降解调节KEAP 1的水平,受损的自噬导致SQSTM 1积累,导致KEAP 1降解和NFE 2L 2蛋白稳定性增加。在细胞核中,NFE 2L 2与靶基因启动子区域中的称为抗氧化反应元件/战神或亲电反应元件/EpRE的特异性DNA序列结合。这种结合激活了一组参与GPX 4依赖性和GPX 4非依赖性途径的基因的转录,以抑制铁中毒。一个关键的未回答的问题是NFE 2L 2如何选择性地激活靶基因来抑制铁凋亡而不是其他类型的细胞死亡。
TP53:TP53在调节铁凋亡易感性方面具有双重作用。例如,乙酰化缺陷型TP53变体TP53[3KR]缺乏诱导细胞凋亡和细胞周期停滞的能力。然而,它通过抑制SLC7A11表达保留了与野生型TP53相似的肿瘤抑制能力,从而增加了某些癌细胞中的铁凋亡敏感性。TP53介导的VKORC1L1下调还通过维生素K代谢增加癌细胞中的铁凋亡敏感性。此外,TP53 通过诱导 SAT1 的表达正向调节铁死亡,SAT1 是多胺分解代谢中的一种限速酶,可产生 ROS。相反,在某些条件下,TP 53抑制铁凋亡。例如,在人类结肠直肠癌细胞中,TP 53缺失通过激活细胞膜上的DPP 4 NOX 1途径增加了对erastin触发的铁凋亡的敏感性。经典的TP 53诱导基因CDKN 1A/p21也抑制癌细胞中的铁凋亡。此外,TP 53突变(R175 H)产生了一种修饰的TP 53蛋白,其通过阻止BACH 1介导的SLC 7A 11下调,从而促进肿瘤生长,起到铁凋亡抑制剂的作用。这些发现强调了TP 53在铁凋亡调节中的广泛意义。
ATF 4:ATF 4(转录激活因子4)在内质网应激和氨基酸代谢中起着至关重要的作用。ER应激激活的ATF 4上调抗铁蛋白基因,如HSPA 5、SLC 7A 11或TXNDC 12。这种途径可以防止癌细胞和线粒体心肌病中的铁凋亡。由促凋亡BH 3模拟物(ABT-737和S63845)诱导的亚致死细胞色素c释放可导致癌细胞中的ATF 4依赖性化疗抗性。考虑到ER作为铁凋亡关键细胞器的重要性,ATF 4可能在转录调节、保持细胞活力和赋予铁凋亡抗性中发挥特定作用。
其他重要的转录因子,包括HIF 1A、NF-κ B/NF-κ B、YAP 1、WWTR 1和SREBF 1,也通过多个靶基因在形成铁蛋白应答中发挥环境依赖性作用。
膜修复系统
Ca 2+是膜修复反应的关键启动剂。当质膜受损时,Ca 2+从外部来源进入细胞质,向下游修复过程发出信号,例如转运所需的内体分选复合物(ESCRT)-III 和胞吐,从而增强铁凋亡抗性。有效的膜修复对细胞功能至关重要,并且其破坏可能是不可逆的。然而,来自不同细胞器的Ca 2+信号在控制铁凋亡敏感性方面具有双重作用,强调了及时监测的重要性。治疗机会和挑战
治疗机会
临床前研究表明,靶向铁凋亡对各种疾病具有广泛的影响,特别是在肿瘤学、神经退行性疾病和I/R损伤中,如下所述。
癌细胞经常发生代谢变化,破坏氧化还原平衡,增加它们对抗氧化剂的依赖,使它们容易受到铁凋亡诱导。靶向铁凋亡提供了一种克服治疗限制的新方法,尽管偶尔存在耐药机制(例如,由于嘧啶或过氧化氢的生物合成增强)。此外,某些实体癌中KRAS和TP 53等基因的特定突变与铁凋亡敏感性相关,为精准医学策略提供了潜力。
神经退行性疾病,如阿尔茨海默病、帕金森病和亨廷顿病,涉及脑中的神经元破坏和蛋白质聚集。氧化应激在这种变性中起关键作用,导致脂质过氧化和铁凋亡细胞死亡。靶向铁凋亡抑制的治疗旨在减少氧化损伤并提高神经元存活率。调节铁凋亡途径可能有助于减轻有害副产物的积累,如脂质过氧化物和反应性醛,可能减缓神经变性,包括多发性硬化症等疾病。
I/R事件引发氧化应激和细胞死亡,使得针对铁氰化钾的治疗有望在中风、心肌梗死以及肾脏和肝脏损伤等疾病中减轻氧化损伤并保护组织功能。结合铁凋亡和坏死性凋亡抑制已显示出特别的效果。对于肾小管,铁凋亡细胞死亡传播遵循被称为“死亡波”的独特模式,并且此后也在其他系统中描述。这些研究强调了铁凋亡抑制剂在I/R相关疾病中的治疗潜力。
治疗挑战
特异性和选择性:需要高特异性和选择性,以最大限度地减少脱靶效应和潜在毒性。例如,人们担心RSL3和ML162对TXNRD 1蛋白的脱靶效应。咪唑酮erastin(IKE)是一种广泛应用的体内铁凋亡诱导剂,但其相对于其他体外激活剂的活性尚需进一步研究。此外,通过抗氧化机制抑制铁凋亡可能会影响非铁凋亡途径,包括凋亡和坏死性凋亡。
药物递送:开发靶向药物递送系统对于提高治疗效果和减少全身副作用至关重要。最近的研究表明,使用纳米颗粒,包括脂质体、胶束和基于聚合物的载体,来解决这些挑战是有希望的。纳米颗粒提供了诸如增强的药物稳定性,溶解性和靶向递送的优点。
生物标志物鉴定:已经在mRNA或蛋白质水平上测量了几种生物标志物,例如TFRC、ACSL 4和PTGS 2、超氧化PRDX 3,以监测铁蛋白沉积反应。从理论上讲,基于血液的生物标志物具有很强的临床应用转化潜力,特别是危险信号,如HMGB 1、ATP、SQSTM 1和DCN(核心蛋白聚糖),它们可以指示铁细胞增多症期间的质膜破裂。DCN以其区分铁细胞凋亡与其他细胞死亡类型的能力而著称,尤其是在早期阶段。基于LC-MS的氧化还原脂质组学是表征体内,尤其是在各种疾病条件下的铁蛋白生物标志物的有价值的工具。
副作用:目前广泛使用的铁凋亡激活剂缺乏细胞或组织选择性,可能导致各种免疫细胞类型(如嗜铁细胞、CD8+ T细胞、自然杀伤细胞和树突细胞)的意外细胞死亡。需要策略来选择性地靶向肿瘤细胞,同时保持免疫细胞的完整性和抗癌免疫应答。一种名为 N6F11 的化合物通过触发 TRIM25 依赖性 GPX4 降解,在癌细胞(而非免疫细胞)中选择性诱导铁死亡方面显示出前景。铁缺乏症治疗还可导致不良反应,如早发性恶病质、干细胞死亡、骨髓损伤、造血破坏和炎症驱动的肿瘤发生。
临床转化:虽然一些FDA批准的药物如索拉非尼、柳氮磺胺吡啶、青蒿琥酯和扎西他滨已显示出诱导临床前铁凋亡的潜力,但它们的作用可能与不良脱靶效应有关。为患者确定安全的药物至关重要,因为考虑联合用药以减轻全身毒性,并探索间歇性治疗方案以提高耐受性。未来的研究应针对这些方面来了解人类疾病中的铁下垂。设计良好的临床试验对于评估铁中毒靶向药物的有效性、安全性和长期结局至关重要。这些试验应招募特定的患者人群,识别敏感的铁下垂症生物标志物,并在临床结局的同时测量它们。
结论与展望
近年来,铁垂症的研究领域出现了显著的增长。这一激增反映了一个真正的以铁中毒为重点的研究时代的建立。然而,最初的定义铁细胞增多症的Fe(II)依赖性调节坏死伴脂质过氧化作用,现在被认为是不完整的。尽管铁诱导的氧化应激仍然是一个突出的触发因素,但其他非铁依赖性刺激或应激无疑与铁下垂症有关。考虑到铁细胞凋亡的核心下游特征是由不受控制的脂质过氧化作用导致的细胞膜结构性损伤,术语“脂毒性”也可能反映了其核心机制。
铁蛋白缺乏症的分子机制已经超出了最初的GPX4调节途径。这篇综述探讨了促铁蛋白和抗铁蛋白机制之间的相互作用,分为GPX4依赖性和GPX4独立性,包括历史见解和最近的研究结果。然而,关于这些通路何时、何地以及如何激活的问题仍然存在。
许多与铁细胞凋亡相关的调控分子也在其他类型的细胞死亡中发挥作用,强调了细胞间串扰的复杂性。解开这些机制需要精心设计的实验、严格的控制和特定生物标志物的验证。了解生理和病理应激因素如何在现实世界中影响铁垂症仍然是一个挑战。此外,导致铁性和非铁性细胞死亡的应激途径之间的复杂联系需要进一步阐明。
尽管偶尔的研究限制和相互矛盾的假设,我们保持乐观的未来前景ferroptosis。我们相信,铁下垂的原则将最终找到超出其启发价值的临床应用。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号