

Hi-C还原我们染色体的真实构想
转自:https://www.sohu.com/a/225120691_99971433
1. 什么是Hi-C技术?
2009年,Job Dekker 研究团队利用Hi-C技术通过测量人类正常淋巴细胞染色体中基因座空间交互信息,首次提出Hi-C技术的概念。
Hi-C技术——以高通量测序为手段,结合生物信息分析方法,以3C (Chromosome Conformation Capture)技术为基础的染色质构象捕捉技术,研究全基因组范围内整个染色质DNA在空间位置上的关系,获得高分辨率的染色质三维结构信息。Hi-C技术不仅可以研究染色体片段之间的相互作用,建立基因组折叠模型,还可以应用于基因组组装、单体型图谱构建、辅助宏基因组组装等,并可以与RNA-seq、ChIP-seq等数据进行联合分析,从基因调控网络和表观遗传网络来阐述生物体性状形成的相关机制。
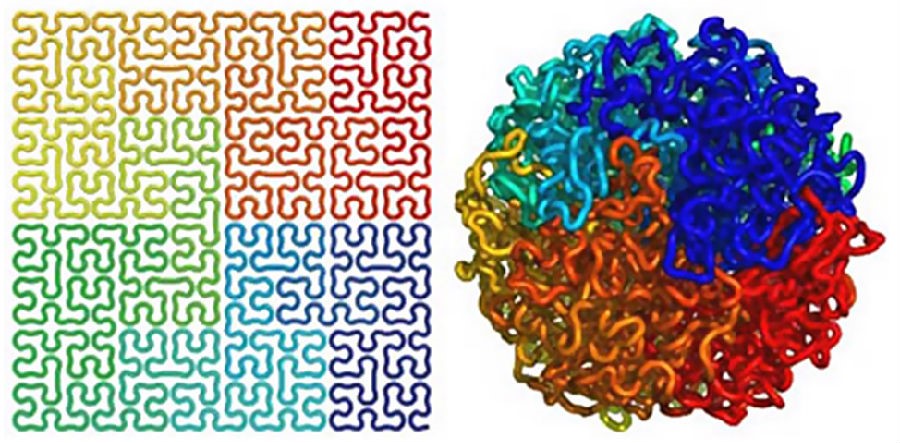
染色体的3D构象
2. 为什么Hi-C技术那么火?
Hi-C技术之所以火了,应该归因于三维基因组学的热度。结构基因组测序得到了很多物种的基因组信息,需要对基因组进行精细结构的注释,解释基因组功能的复杂性和多样性。很多基因在染色体和染色质不同状态转换时(超螺旋、复杂折叠等),在空间上的距离不断发生变化。这些基因彼此之间如何调控、如何相互影响?是一个非常有吸引力的课题,需要大量的研究来探讨。Hi-C技术可以基于高通量进行染色体构象的捕获,它能够在全基因组范围内捕捉不同基因座位之间的空间交互,研究三维空间中调控基因的DNA元件。因此Hi-C技术火起来也是情理之中的事情。开创性的研究才能发高分文章哦~
3. 怎么应用Hi-C技术?
知道了,Hi-C技术那么火!那怎么应用这个技术到我们的科研中呢?或者说有哪些研究可以用这个技术呢?Hi-C有很多用武之地,比如说,(1)探索基因组的3D结构,有助于了解基因组折叠对基因的表达和调控的影响;(2)开发调控基因的DNA元件,揭示基因组远程调控元件介导的分子网络;(3)构建染色质跨度的单体型图谱:为肿瘤形成机制提供依据、为疾病风险预测提供思路、为农业动植物经济性状连锁标记及基因组进化奠定基础;(4)可以与RNA-Seq、ChIP-Seq等数据进行联合分析,从基因调控网络和表观遗传网络来阐述生物体性状形成的相关机制。

4. Hi-C相关的高质量SCI文章发!发!发!(重要的事情说三遍)
这么火的技术,从2009年推出以来,一直有高质量SCI文章发表,越来越多的研究利用Hi-C技术揭秘不同物种染色体的空间构象和调控机制。详细文章见下表:
基于Hi-C技术发表的部分文章列表
发表作者 发表时间 研究内容 发表期刊 研究物种
Lieberman-Aiden et al. 2009 人淋巴细胞染色体中 Science 人
基因座空间交互信息
Sexton et al. 2012 构建果蝇分辨率的 Cell 果蝇
三维基因组互作图谱
Dixon et al. 2012 人及小鼠胚胎干细胞 Nature 人和小鼠
的三维基因结构
Rao et al. 2014 绘制人基因组详细的 Cell 人
图谱
Chandra et al. 2015 衰老细胞的细胞核 Cell Reports 人
空间结构
Mifsud et al. 2015 基因组启动子的 Nature Genetics 人
空间互作
Barutcu et al. 2015 乳腺癌细胞染色体 Genome Biology 人
三维结构特征
Manuel et al. 2017 心力衰竭细胞系高分 Circulation 人
辨率染色体构象解析
5. 如何分析Hi-C数据?
虽然Hi-C的优势在于其结合了二代测序,增加了数据分析结果的完整性,但是,这势必也使得其数据分析相对复杂了。想学习如何分析Hi-C数据?可以阅读生信草堂之前的推文:功能基因组学研究利器——Hi-C这篇推文详细介绍了Hi-C技术的:工作流程+生物信息学分析方法+数据的可视化。
6. 文献解读:如何利用Hi-C技术研究乳腺癌发生机制?
这里我们给大家分享一篇发表在《Genome Biology》上的文章:“Chromatin interaction analysis reveals changes in small chromosome and telomere clustering between epithelial and breast cancer cells”。这篇文章利用Hi-C技术研究乳腺癌细胞染色体三维结构特征。
本文的样本选择是人乳腺上皮细胞系MCF-10A和乳腺癌细胞系MCF-7。利用的文库和测序策略是Hi-C文库(HiSeq 2000测序,PE100)+转录组文库(HiSeq 2000测序,SE100)。
首先,通过进行1M分辨率下人乳腺上皮细胞系MCF-10A和乳腺癌细胞系MCF-7在染色体互作的差异分析,结果如下图:

 通过染色体相互作用热图比较,肉眼很难发现它们之间的差异。本文采用“减法”算法,巧妙地将两个细胞系的互作热图整合到一张图中,发现1M分辨率下人乳腺上皮细胞系MCF-10A和乳腺癌细胞系MCF-7在染色体互作上存在差异(如上图)。发现在16-22号染色体中,MCF-10A细胞系中有较强的基因相互作用(如下图)。
通过染色体相互作用热图比较,肉眼很难发现它们之间的差异。本文采用“减法”算法,巧妙地将两个细胞系的互作热图整合到一张图中,发现1M分辨率下人乳腺上皮细胞系MCF-10A和乳腺癌细胞系MCF-7在染色体互作上存在差异(如上图)。发现在16-22号染色体中,MCF-10A细胞系中有较强的基因相互作用(如下图)。

此前的研究表明,基因组内的相互作用有两种模式:开放式(A-type)和封闭式(B-type)。在人乳腺上皮细胞系MCF-10A和乳腺癌细胞系MCF-7基因组中鉴定出了两种区室,MCF-10A和MCF-7的开方式和封闭式区域分布大体相似,只有一些区域有A到B区室或者B到A区室的转化。这些转化区域中的很多基因和癌症重要通路WNT相关(如下图)。

MCF-10A和MCF-7基因组中大规模染色体相互作用差异和改变的基因区室内是否会对TAD(Topologically associating domains;这里TAD被定义为相互调节的基因集,包含的基因的增强子和启动子之间相互作用)形成和基因表达造成影响是未知的。为解决这个问题,用40kb分辨率来鉴定TAD边界,发现一些TAD边界是乳腺癌细胞系特有的(如下图)。

转自生信草堂
生信草堂
浙大生信博士团队倾力打造的一个科研人员学习交流的公众微信平台。我们致力于科研社区服务,分享最前沿的科技进展,提供生信分析方法,解读经典分析案例,公众数据库的挖掘和临床数据统计分析。在此我们欢迎各位的加入!

 浙公网安备 33010602011771号
浙公网安备 33010602011771号