
易基因:Adv Sci/IF14.1:ChIP-seq+ATAC-seq等揭示进化保守的TPM1超级增强子调控骨骼肌发育的表观新机制
大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。
近日,广西大学张瑞门助理教授为第一作者,韦英明、杨素芳、邓彦飞教授为共同通讯作者,在国际知名期刊《Advanced Science》(IF14.1/Q1)上发表题为“The Evolutionarily Conserved TPM1 Super-Enhancer Drives Skeletal Muscle Regeneration via Mechanotransduction Signaling”的科研成果。研究通过牛、小鼠、人的多物种ChIP-seq、ATAC-seq表观基因组学联合分析,首次鉴定出一个在进化上高度保守的超级增强子TPM1_SE(TPM1 super-enhancer),系统揭示了TPM1_SE通过整合表观遗传调控与细胞信号协同促进骨骼肌再生与发育的全新机制。
本研究结果表明,位于编码肌动蛋白稳定蛋白的TPM1_SE超级增强子,通过转录因子TEAD4介导的染色质环化机制,在种属间呈现差异化转录调控模式:在小鼠中促进线性TPM1 mRNA表达,而在牛中则特异性调控环状RNA CircTPM1生物合成。研究系统阐明了TPM1_SE通过激活PI3K/AKT信号通路和MYH10/MYL3机械信号轴,整合表观遗传调控与细胞信号,从而促进骨骼肌再生与肌管生成的分子机制。该发现不仅为理解超级增强子与细胞互作提供了新思路,也为人类肌肉退行性疾病的治疗及表观遗传育种改良畜禽产肉性能提供了关键分子靶点。

英文标题:The Evolutionarily Conserved TPM1 Super-Enhancer Drives Skeletal Muscle Regeneration via Mechanotransduction Signaling
中文标题:进化保守的TPM1超级增强子通过机械力学信号驱动骨骼肌再生
发表时间:2025年11月16日
发表期刊:Advanced Science
影响因子:IF14.1/Q1
技术平台:ChIP-seq、ATAC-seq等
作者单位:广西大学
DOI: 10.1002/advs.202514271
超级增强子(SEs)是组织再生的关键表观遗传调控因子,然而其在肌肉生成过程与细胞互作的机制尚未完全阐明。本研究发现,进化上保守的超级增强子(TPM1_SE)可能整合表观遗传调控与细胞信号发挥作用。
在体外实验中,TPM1_SE缺失会破坏肌肉生成并降低TPM1及其circRNA亚型CircTPM1表达,条件性敲除TPM1_SE显著减少肌肉质量并延缓再生进程。具体而言,TPM1_SE通过TEAD4介导的染色质环化促进线性TPM1 mRNA(小鼠)和CircTPM1(牛)表达,并与NKX2.2互作激活对细胞机械力敏感的PI3K/AKT信号通路,进而协同调控肌管生成过程中的细胞骨架(Cytoskeleton)重构,而TPM1_SE缺失会破坏NKX2.2-PI3K/AKT信号。
总之,本研究确立了TPM1_SE作为整合表观遗传调控与生物出核的进化保守性枢纽。小鼠模型揭示了其在肌肉再生医学中的治疗潜力,而牛CircTPM1介导的机制则突显了TPM1_SE作为改良家畜肉品质遗传性状的潜在靶点。
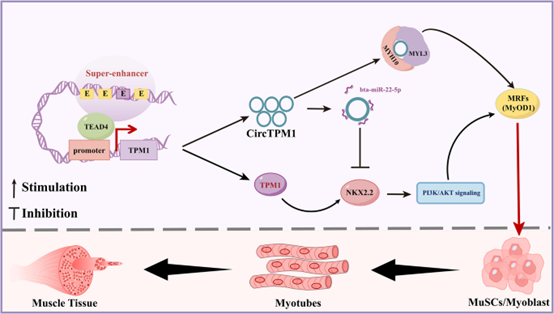
图形摘要:TPM1_SE激活TPM1位点,增强PI3K/AKT信号通路和MYH10/MYL3轴活性,从而促进骨骼肌再生。具体而言,TPM1_SE通过TEAD4介导的染色质环化作用激活TPM1转录并促进CircTPM1形成,进而增强PI3K/AKT信号及MYH10/MYL3活性,在骨骼肌再生过程中调控肌管生成的细胞骨架重构。
易小结
本研究突破了传统表观遗传学与细胞互作研究界限,具有明确的双向转化潜力。一方面,为干预衰老性或病理性肌肉退行性疾病提供新靶点;另一方面,为动物农业中通过精准分子育种实现“优质高产”提供了原创性靶标和理论支撑,有望带来经济效益。
本研究是ChIP-seq、ATAC-seq等分子机制研究技术与细胞、小鼠模型、力学等功能验证相结合的典型示例,为研究复杂生物学过程中的非编码调控元件提供了方法学参考。
研究方法
(1)表观基因组学测序分析
染色质免疫共沉淀测序(ChIP-seq):对牛肌肉干细胞(MuSCs)在增殖期(GM)和分化期(DM)的组蛋白修饰(H3K4me1, H3K27ac)进行测序,结合人骨骼肌成肌细胞(GEO数据集)及小鼠C2C12成肌细胞(GEO数据集),在全基因组范围内鉴定和表征超级增强子(SEs)。
染色质可及性测序(ATAC-seq):分析牛MuSCs和小鼠C2C12细胞的染色质可及性,鉴定活跃的顺式调控元件(CREs)和TEAD4等关键调控因子的结合位点。
数据整合与比较:结合已公开的人类和小鼠肌肉细胞的ChIP-seq/ATAC-seq数据(GEO数据库),进行跨物种比较,鉴定进化保守的SEs及其靶基因。
(2)功能基因组学与分子生物学验证:
报告基因实验:将预测的增强子元件克隆到荧光素酶报告载体中,验证其转录活性。
ChIP-qPCR:针对候选因子(如TEAD4)和组蛋白修饰(H3K27ac),验证其在特定基因组位点(如TPM1_SE和TPM1启动子)的结合或富集情况。
3C-qPCR实验:直接证实TPM1_SE(bCRE_9/mCRE_26)与TPM1启动子之间存在物理互作的染色质环,从三维基因组层面揭示调控机制。
基因编辑:利用CRISPR/Cas9技术在细胞(C2C12,牛MuSCs)和小鼠(条件性敲除mCRE_26)中敲除TPM1_SE核心元件,研究其功能缺失表型。
过表达/敲低:在细胞中过表达或敲低TPM1、CircTPM1、TEAD4等分子,研究其功能获得表型。
(3)转录组与RNA生物学:
circRNA-seq:对牛MuSCs的GM/DM样本进行测序,系统鉴定差异表达的circRNA,并从中发现了受TPM1_SE调控的CircTPM1。
RT-qPCR:定量检测各种RNA分子(mRNA/circRNA/miRNA)的表达水平。
RNA免疫共沉淀(RIP):验证CircTPM1与MYH10蛋白的直接结合。
染色质分离RNA纯化(ChIRP):验证CircTPM1与bta-miR-22-5p的互作。
FISH:观察CircTPM1与bta-miR-22-5p或MYH10的亚细胞共定位。
(4)蛋白水平与互作研究:
Western Blot:检测关键信号通路蛋白(PI3K, AKT)和肌源性标志蛋白(MyOD1, MyHC)表达。
免疫共沉淀(Co-IP):验证蛋白质间互作,如NKX2.2与PI3K互作,MYH10与MYL3互作。
(5)细胞与动物模型表型分析:
细胞机械力学表征:检测细胞在流体应力下的形变,定量敲除或过载TPM1_SE下游分子对细胞刚度的影响,将分子功能与细胞机械力学特性直接关联。
小鼠模型:构建肌肉特异性TPM1_SE敲除鼠(mCRE_26cKO),系统监测其生长、肌肉重量、以及心脏毒素(CTX)诱导损伤后的肌肉再生能力。
结果图形
(1)肌肉生成过程中保守的超级增强子(SEs)与TPM1位点的染色质互作
研究团队通过对牛、人、小鼠肌肉细胞的ChIP-seq和ATAC-seq数据分析,首次在三个物种的TPM1位点鉴定出一个进化保守的、富含H3K27ac修饰的超级增强子,命名为TPM1_SE(图1D-F),该SE由多个顺式调控元件(CREs)组成。牛物种的双荧光素酶报告实验筛选出活性最强的元件bCRE_9(图1G),ChIP-qPCR证实其H3K27ac富集水平最高(图1H)。小鼠物种的同源元件mCRE_26也展现出强增强子活性(图1I-J)。最关键的是,利用染色体构象捕获(3C)技术研究证实了bCRE_9/mCRE_26与TPM1启动子之间存在特异的物理互作,形成了染色质环(图1K-L)。从三维基因组结构上证明了TPM1_SE能够直接调控TPM1基因转录,为后续功能研究奠定了基础。
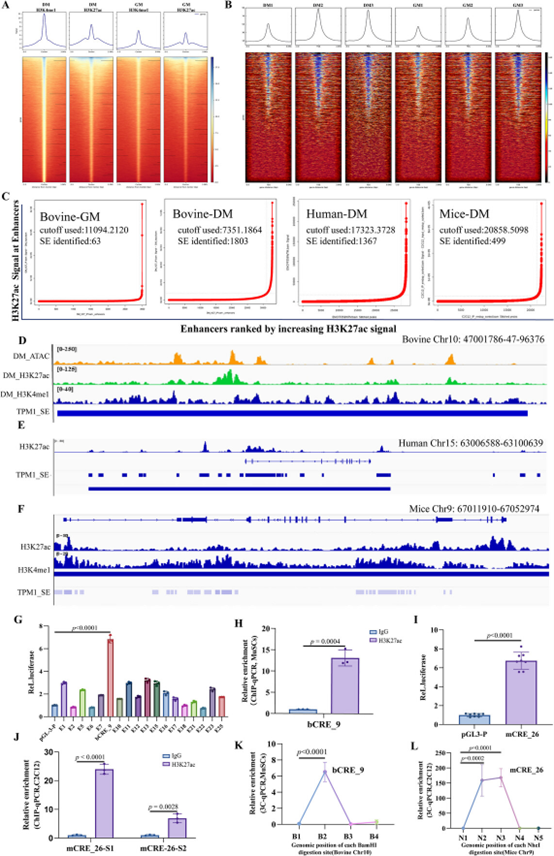
图1:肌肉生成过程中TPM1位点的保守超级增强子及染色质互作
A) 基于牛肌肉干细胞(MuSCs)的ChIP-seq数据,对增殖期(GM)和分化期(DM)样本的H3K27ac和H3K4me1信号进行差异分析,对所有SEs进行排序并绘制其在基因转录起始位点(TSSs)附近的分布图谱。
B) 基于ATAC-seq的染色质可及性图谱生成线图和热图,展示GM和DM样本中TSS位点的reads分布。结果显示所有六个样本在TSS附近均表现出富集的reads分布,且越接近TSS,富集强度显著增加。
C) 牛MuSCs-GM/DM、人骨骼肌成肌细胞-DM和小鼠C2C12成肌细胞-DM中特异性与增殖或肌肉生成相关的超级增强子的定量分析。
D) IGV可视化牛TPM1_SE区域的活性组蛋白标记和ATAC信号peaks。
E-F) 在IGV中注释活性组蛋白修饰展示人(E)和小鼠(F)基因组中TPM1_SE的比较染色质图谱。
G) 双荧光素酶报告基因实验验证牛TPM1_SE中各顺式调控元件的转录活性。
H) ChIP-qPCR评估bCRE_9区域的H3K27ac富集水平。
I-J) 分别通过荧光素酶报告实验(I)和ChIP-qPCR(J)评估mCRE_26的转录活性及H3K27ac占据情况。
K-L) 3C-qPCR实验揭示特定引物与检测引物之间的互作信号,证实TPM1_SE与TPM1启动子之间存在物理互作。
(2)TPM1_SE缺失损害肌肉生成及TPM1位点的转录活性
敲除TPM1_SE核心元件(mCRE_26-KO C2C12或bCRE_9-KO 牛MuSCs)后,肌肉生成严重受损,表现为MyHC阳性肌管数量减少、融合指数下降、肌源性标志物表达降低(图2A-H)。RT-DC力学检测显示,敲除细胞变形度增加,表明细胞刚度下降(图2I-J),提示TPM1_SE参与调节细胞机械力学特性。小鼠C2C12细胞中敲除mCRE_26导致TPM1 mRNA表达水平显著下调(图2K),而在牛MuSCs中敲除bCRE_9却不影响TPM1 mRNA(图2L)。这一差异引导研究者转向非编码RNA。
通过对牛MuSCs的circRNA-seq分析,研究团队鉴定出一个由TPM1基因产生的658-nt环状RNA——CircTPM1,其表达在肌肉生成过程中上调(图2M、N、P)。使用SE功能抑制剂JQ1处理或直接敲除bCRE_9,都能显著降低CircTPM1表达,而不影响线性mRNA(图2Q-S)。上述研究结果首次揭示了TPM1_SE在物种间调控出核分化:在小鼠中主要调控线性TPM1 mRNA,而在牛中特异性调控环状RNA CircTPM1,证明同一SE可通过物种差异性的转录出核(小鼠mRNA vs 牛circRNA)调控肌肉生成。
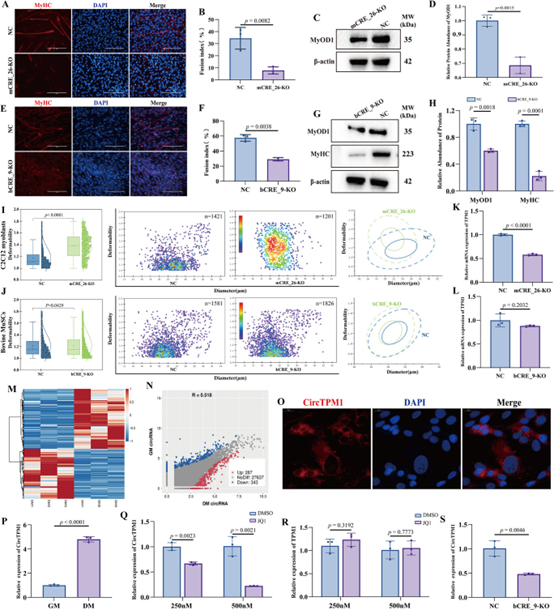
图2:TPM1_SE缺失损害肌肉生成和TPM1转录
A) 免疫荧光分析显示,mCRE_26缺失的C2C12成肌细胞中MyHC阳性肌管形成减少。
B) 融合指数计算为MyHC阳性肌管中的细胞核百分比。
C-D) WB分析和定量证实MyOD1蛋白水平的下调。
E) 在牛MuSCs中,bCRE_9缺失同样损害肌肉生成,表现为MyHC阳性肌管减少。
F) 融合指数。
G-H) WB和光密度分析显示MyOD1和MyHC蛋白表达降低。
I-J) 在mCRE_26敲除(KO)C2C12成肌细胞(I)和bCRE_9敲除牛MuSCs(J)中评估细胞变形性。
K-L) RT-qPCR显示CRE元件缺失显著降低了C2C12成肌细胞中TPM1 mRNA水平,但不影响牛MuSCs中的水平。
M-N) 牛MuSCs中GM期和DM期差异表达circRNA热图,鉴定出630个显著表达变化circRNA。
O) FISH分析显示CircTPM1主要定位于牛MuSCs的细胞质中。
(3)TPM1_SE缺失降低肌肉重量并延缓骨骼肌再生
为在体内验证TPM1_SE的功能,研究团队构建了mCRE_26肌肉特异性条件敲除小鼠(mCRE_26cKO; Myf5-Cre)。研究结果显示,与对照组相比,cKO小鼠从3周龄起体重显著降低,8周龄时腓肠肌(GAS)、胫骨前肌(TA)、比目鱼肌(SOL)和股四头肌(QUA)重量分别减少2.81%、5.07%、34.34%和3.03%(图3A-E)。组织学分析显示TA肌纤维横截面积分布左移,提示肌纤维生长受阻(图3F)。
更重要的是,在CTX诱导的肌肉损伤再生模型中,mCRE_26cKO小鼠表现出严重的再生缺陷:损伤后TA肌肉的重量/长度比恢复更差(图3H-K),肌纤维排列松散、密度和面积减小,再生肌纤维比例降低(图3L-Q),同时肌肉干细胞标记物Pax7和肌肉生成标记物(MyoD1, MyoG, MyHC)的表达持续低下(图3R, S)。这些体内实验强有力地证明,TPM1_SE对于维持正常的肌肉生长和损伤后的高效再生必不可少。
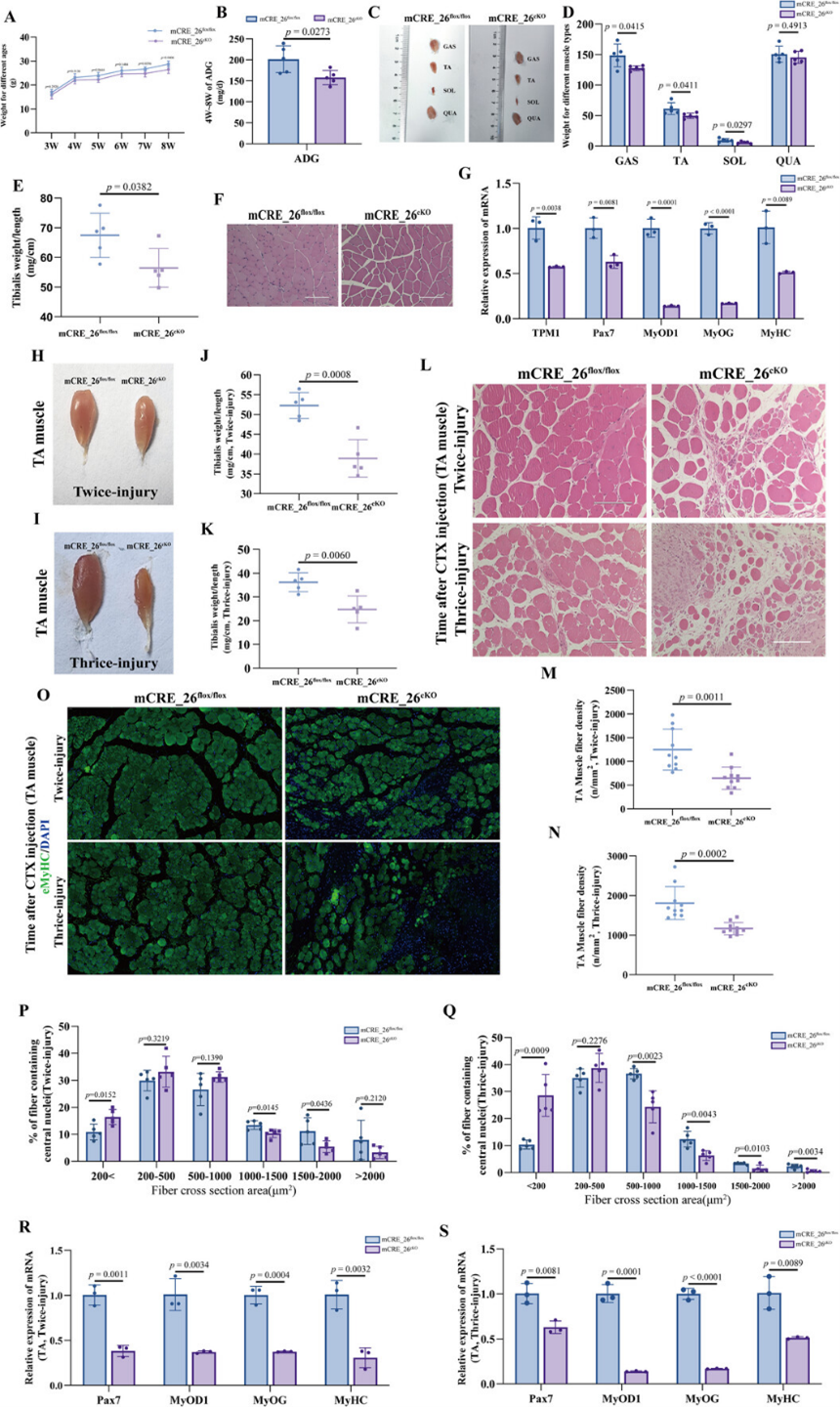
图3:TPM1_SE缺失会减少肌肉质量并延缓骨骼肌再生。
(4)TPM1_SE通过TEAD4介导的染色质互作调控TPM1位点转录
为揭示TPM1_SE发挥功能的分子机制,研究者对ATAC-seq数据进行motif分析,预测并验证了转录因子TEAD4在TPM1_SE(bCRE_9/mCRE_26)和TPM1启动子上均有结合位点(图4A-D、P-R)。ChIP-qPCR证实TEAD4在这些位点的富集(图4C-D、Q-R)。报告基因实验显示,TEAD4过表达能显著增强由TPM1_SE和TPM1启动子介导的转录活性,而突变TEAD4结合位点则使此激活效应消失(图4E、S)。
具体而言,在牛MuSCs中过表达TEAD4能上调CircTPM1并促进分化(图4G、I-L),敲低TEAD4则抑制分化(图4M-O);在小鼠C2C12细胞中过表达TEAD4上调TPM1 mRNA(图4U)。这些结果表明,TEAD4是介导TPM1_SE与TPM1启动子形成染色质环并激活转录的关键因子(图4F、T),构成该保守调控模块的核心。
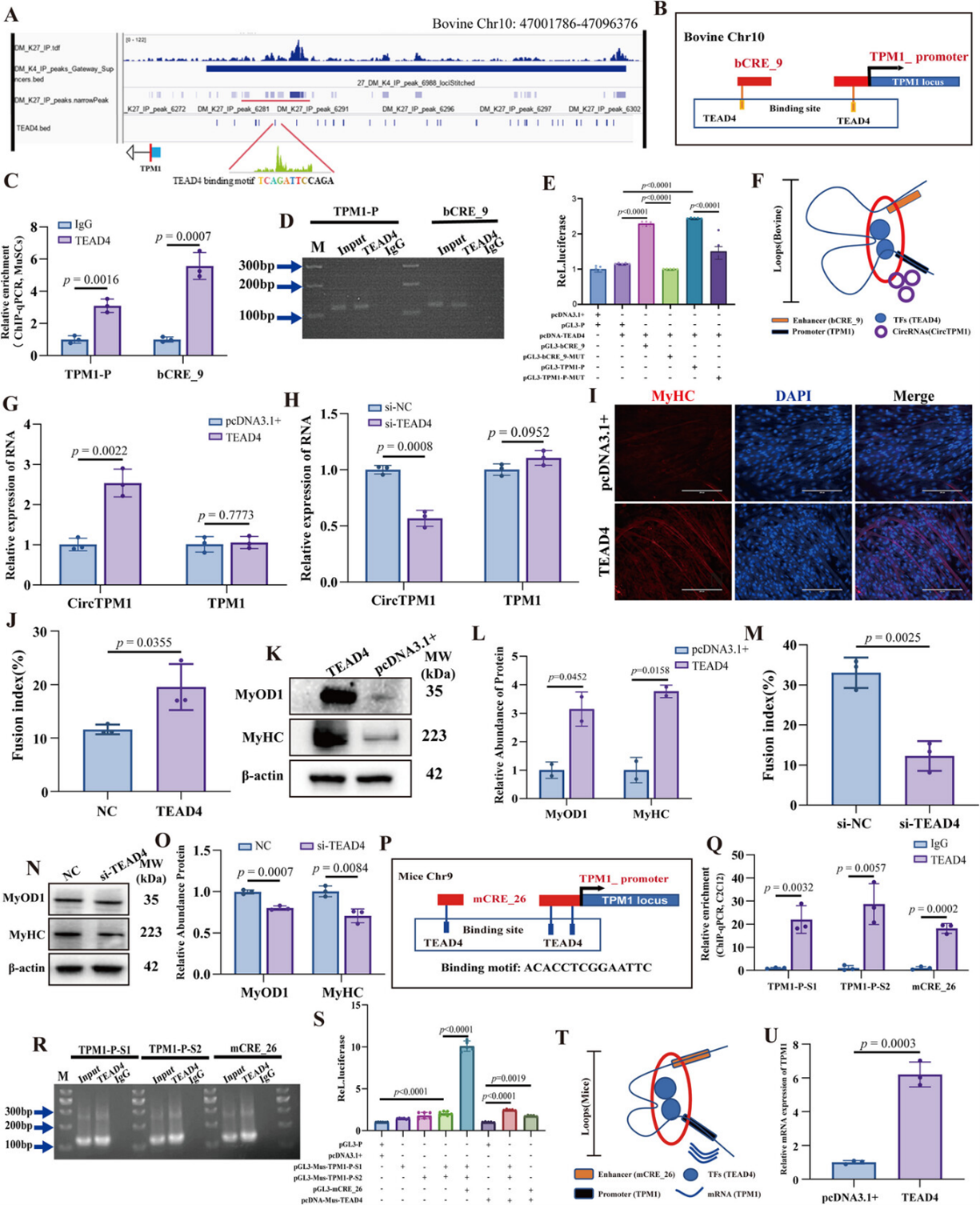
图4:TPM1_SE通过TEAD4介导的染色质环化调控TPM1转录
(5)TPM1来源的RNA转录本促进肌肉生成并增强骨骼肌再生
为验证TPM1转录本的功能,研究团队进行了物种特异性过表达实验:在小鼠C2C12细胞中过表达线性TPM1 mRNA,在牛MuSCs中过表达CircTPM1。结果显示两种转录本均显著促进多核肌管生成,提高MyOD1和MyHC蛋白水平,并增加融合指数(图5A-H)。值得注意的是,CircTPM1在小鼠C2C12细胞中同样具有促分化功能(图5I-L),提示其机制跨物种保守。
RT-DC检测显示,过表达TPM1或CircTPM1的细胞变形性显著降低,表明细胞刚度和弹性模量增加(图5M-N),证实这些转录本通过机械力学信号中发挥作用。在体内实验中,CTX损伤后注射CircTPM1质粒可显著提高TA肌重量/长度比,减少血肿形成,增加肌纤维密度和横截面积,提高再生肌纤维比例(图5O-T)。免疫荧光显示eMyHC+细胞增多(图5U),且内源性小鼠TPM1 mRNA水平也显著上调(图5V)。这些结果证实TPM1来源的转录本通过增强细胞机械力学特性和肌肉生成程序促进组织修复。

图5:TPM1来源的RNA转录本促进肌肉生成与骨骼肌再生
(6)TPM1来源的转录本通过PI3K/AKT信号通路促进肌肉发育
研究进一步探究了下游信号通路。生物信息学分析和分子对接模拟提示TPM1与PI3K/AKT通路相关。实验结果证实,过表达TPM1(小鼠)或CircTPM1(牛)可显著上调NKX2.2及PI3K/AKT通路组分的蛋白水平(图6A-C、G-H)。Co-IP提示NKX2.2与PI3K存在互作(图6D)。反之,敲除TPM1_SE则抑制该通路(图6E-F、S-T)。
具体而言,CircTPM1作为ceRNA(竞争性内源RNA)吸附bta-miR-22-5p,解除对NKX2.2抑制。双荧光素酶报告基因证实CircTPM1与bta-miR-22-5p直接结合(图6I),ChIRP-qPCR和FISH证实二者在细胞质中共定位(图6J-K)。过表达bta-miR-22-5p抑制NKX2.2,而CircTPM1可挽救此效应(图6L)。NKX2.2过表达激活PI3K/AKT并促进分化(图6M-R),而bCRE_9敲除降低NKX2.2和PI3K/AKT(图6S-T)。
重要的是,PI3K/AKT通路激活不仅促进肌管生成,还显著增加细胞刚度(图6W-X),而抑制该通路则导致细胞变软。上述研究结果阐明了TPM1_SE下游转录本通过激活经典且对机械力敏感的PI3K/AKT通路以调控肌肉生成与细胞骨架重构的分子路径。
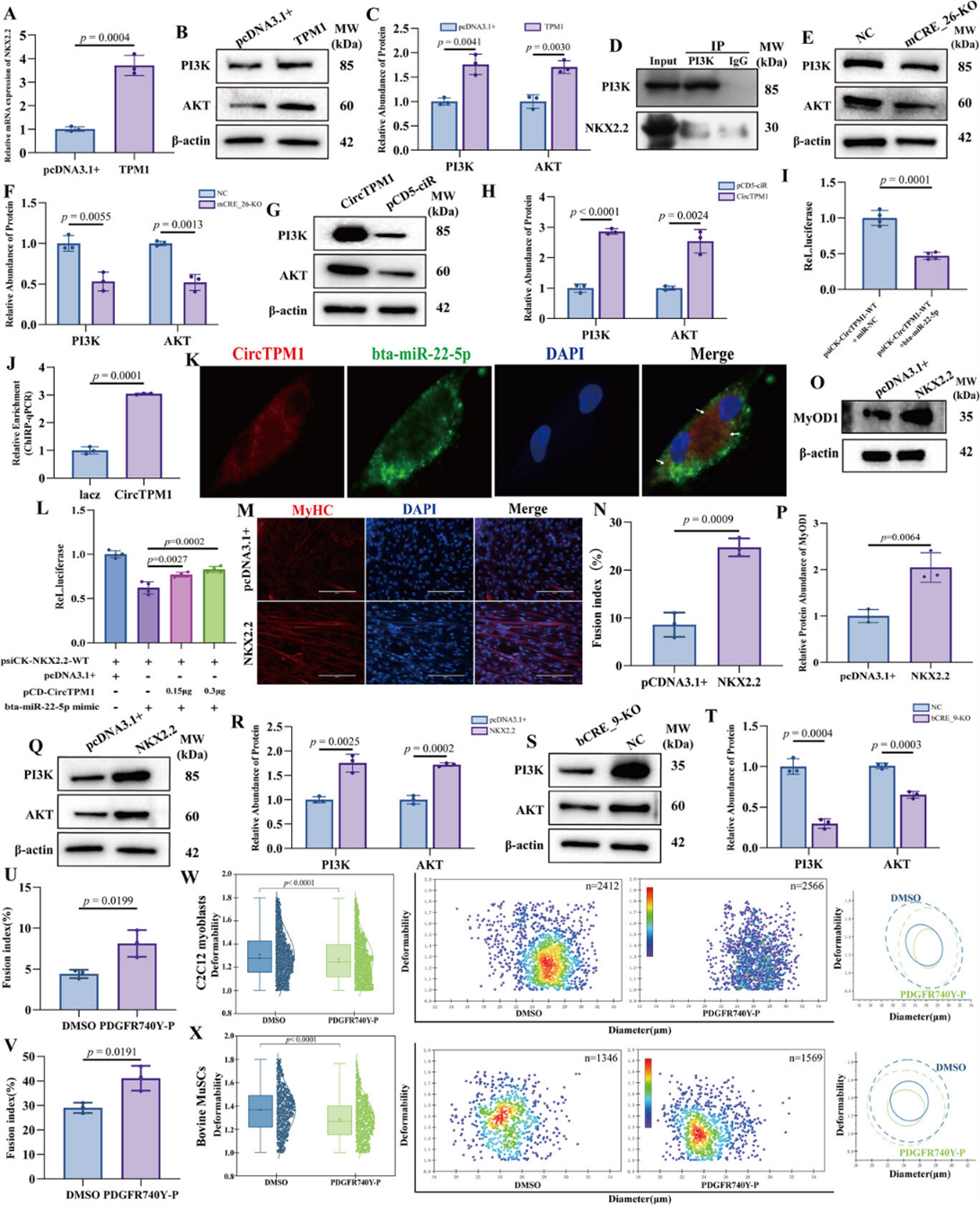
图6:TPM1来源的RNA转录本通过PI3K/AKT信号通路促进肌肉发育。
(7)CircTPM1通过结合MYH10激活MYL3表达以促进肌肉生成
除ceRNA机制外,研究还发现CircTPM1作为RNA结合蛋白(RBP)“海绵”的新功能。通过RIP-qPCR和FISH实验,证实CircTPM1与非肌肉肌球蛋白IIB(MYH10) 直接结合并共定位(图7B、C、G)。过表达CircTPM1能上调MYH10的表达(图7D-F)。进一步的Co-IP结合质谱分析发现,MYH10与肌球蛋白轻链3(MYL3) 存在互作(图7H-I)。
具体而言,MYL3表达在分化时上调,且能被CircTPM1过表达进一步促进(图7J-L);过表达MYL3本身即可有效促进肌管生成(图7M-P)。敲除TPM1_SE则降低MYH10和MYL3表达(图7Q-R)。上述研究结果证实TPM1_SE通过CircTPM1-MYH10/MYL3轴调控肌球蛋白组装和细胞骨架重构。
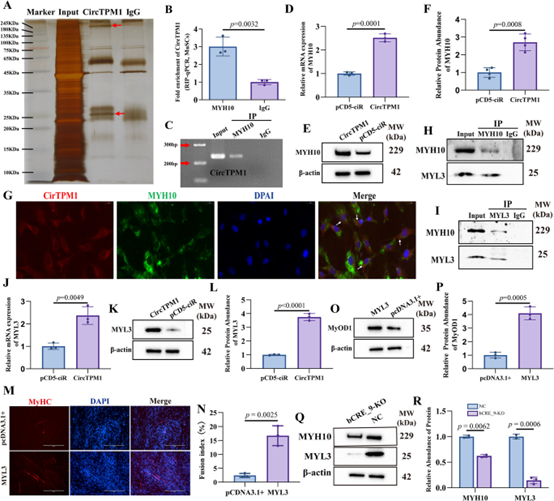
图7:CircTPM1通过直接结合MYH10并上调MYL3表达以促进肌肉生成。
结论和启示
本研究通过ChIP-seq、ATAC-seq、circRNA-seq多组学联合分析揭示了TPM1超级增强子(TPM1_SE)作为进化保守的、整合表观遗传与细胞机械力学的核心纽带,主要通过TEAD4介导的染色质环化,调控TPM1位点产生物种特异性转录本(mRNA/CircTPM1),进而激活PI3K/AKT信号通路和MYH10/MYL3轴,最终促进细胞骨架重构和骨骼肌再生。
ChIP-seq与ATAC-seq在本研究中的核心作用
SE鉴定与定位:ChIP-seq(H3K27ac/H3K4me1)绘制了全基因组SE图谱,在三个物种中同步鉴定TPM1_SE,并确定其由26个CREs组成的核心结构。
染色质可及性状态:ATAC-seq揭示了GM到DM转换过程中的染色质开放区域变化,帮助定位bCRE_9和mCRE_26等关键调控元件,并为3C实验的引物设计提供精确限制性酶切位点信息。
转录因子预测:ATAC-seq数据的motif分析成功预测了TEAD4作为关键调控因子,为后续的机制研究指明方向。
跨物种比较:通过比对牛、人、小鼠的ChIP-seq数据,研究团队确认了TPM1_SE的进化保守性,为其功能重要性提供了进化生物学证据。
参考文献:Zhang R,…et al, Yang S, Deng Y, Wei Y. The Evolutionarily Conserved TPM1 Super-Enhancer Drives Skeletal Muscle Regeneration via Mechanotransduction Signaling. Adv Sci (Weinh). 2025 Nov 16:e14271. doi: 10.1002/advs.202514271.

 浙公网安备 33010602011771号
浙公网安备 33010602011771号