FASTQ格式
FASQT格式是用于存储生物序列(通常是核苷酸序列)及其相应的碱基质量分数的一种文本格式。为简洁起见,序列字母和质量分数均使用单个ASCII字符进行编码。最初由Wellcome Trust Sanger Institute(桑格研究所)开发用于捆绑FASTA格式的序列和其碱基质量分数的,现在已成为存储Illumina Genome Analyzer(Illumina基因组分析仪)等高通量测序仪的标准输出格式。
FASTQ文件格式
- 第1行,以“@” 字符开头,后面跟着一个序列标识符和一个可选的描述(像FASTA格式标题行)。
- 第2行,原始序列字符。
- 第3行,以“+”字符开头,后面可选地跟着相同的序列标识符或者任何描述。
- 第4行,第2行序列每个碱基的质量编码值,包含与第2行字符数量相同的符号。
包含单个序列的FASTQ文件示例如下:
@SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT + !''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
字符“!”和“~”分别代表最低和最高质量,下面是ASCII代表质量值从左到右顺序递增:
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
Illumina序列标识符
来自Illumina软件的序列使用系统标识符:
@HWUSI-EAS100R:6:73:941:1973#0/1
| HWUSI-EAS100R | the unique instrument name |
|---|---|
| 6 | flowcell lane |
| 73 | tile number within the flowcell lane |
| 941 | 'x'-coordinate of the cluster within the tile |
| 1973 | 'y'-coordinate of the cluster within the tile |
| #0 | index number for a multiplexed sample (0 for no indexing) |
| /1 | the member of a pair, /1 or /2 (paired-end or mate-pair reads only) |
Illumina流程1.4版本以后,使用#NNNNNN代替#0作为多重ID,NNNNNN是多重标签的序列。
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
| EAS139 | the unique instrument name |
|---|---|
| 136 | the run id |
| FC706VJ | the flowcell id |
| 2 | flowcell lane |
| 2104 | tile number within the flowcell lane |
| 15343 | 'x'-coordinate of the cluster within the tile |
| 197393 | 'y'-coordinate of the cluster within the tile |
| 1 | the member of a pair, 1 or 2 (paired-end or mate-pair reads only) |
| Y | Y if the read is filtered, N otherwise |
| 18 | 0 when none of the control bits are on, otherwise it is an even number |
| ATCACG | index sequence |
请注意,最新版本的Illumina软件会输出样本编号(从样本表中读取替代)代替索引序列。例如,以下情况可能出现在批次的第一个样本中:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:N:18:1
NCBI SRA数据
来自NCBI/EBI SRA数据库的FASTQ文件通常包含如下一个描述:
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36 GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC +SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36 IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
在本例中,由一个NCBI分配的标识符、描述保存Solexa/Illumina的原始标识符和read长度。测序是在双端模式下进行的(~500bp插入大小),详见SRR001666。值得注意的是,当使用带缺省参数设置的fastq-dump从SRA数据中提取数据时,fastq-dump将正向read和反向read的序列连接在一起,提取的序列具有双倍长度,丢失了双端测序信息。
$ /opt/sratoolkit.2.5.7-centos_linux64/bin/fastq-dump SRR001666 @SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=72 GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACCAAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA +SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=72 IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9ICIIIIIIIIIIIIIIIIIIIIDIIIIIII>IIIIII/ @SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=72 GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGAAGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT +SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=72 IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBIIIIIIIIIIIIIIIIIIIIIIIGII>IIIII-I)8I
更好的方法是保存原始材料并分成两个或三个文件(正向、反向、单个),例如:
$ /opt/sratoolkit.2.5.7-centos_linux64/bin/fastq-dump --origfmt --split-3 SRR001666 $ head SRR001666_1.fastq SRR001666_2.fastq ==> SRR001666_1.fastq <== @071112_SLXA-EAS1_s_7:5:1:817:345 GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC +071112_SLXA-EAS1_s_7:5:1:817:345 IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC @071112_SLXA-EAS1_s_7:5:1:801:338 GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGA +071112_SLXA-EAS1_s_7:5:1:801:338 IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBI ==> SRR001666_2.fastq <== @071112_SLXA-EAS1_s_7:5:1:817:345 AAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA +071112_SLXA-EAS1_s_7:5:1:817:345 IIIIIIIIIIIIIIIIIIIIDIIIIIII>IIIIII/ @071112_SLXA-EAS1_s_7:5:1:801:338 AGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT +071112_SLXA-EAS1_s_7:5:1:801:338 IIIIIIIIIIIIIIIIIIIIIIGII>IIIII-I)8I
另请注意,NCBI已将此FASTQ数据从原始Solexa/Illumina编码转换为Sanger标准(请参阅下面的编码)。
变动
质量
质量值Q是p的整数映射(即相应碱基判定不正确的概率),主要有两种不同的公式被使用。第一种是评估碱基判定的可靠性的不标准Sanger变体,也称为Phred质量分数:

Solexa流程(即与Illumina Genome Analyzer一起交付的软件)较早使用了不同的映射编码概率p/(1-p),而不是p:

虽然两个映射在更高质量值处渐近地相同,但它们在较低质量水平下不同(即,近似p> 0.05等同Q <13)。
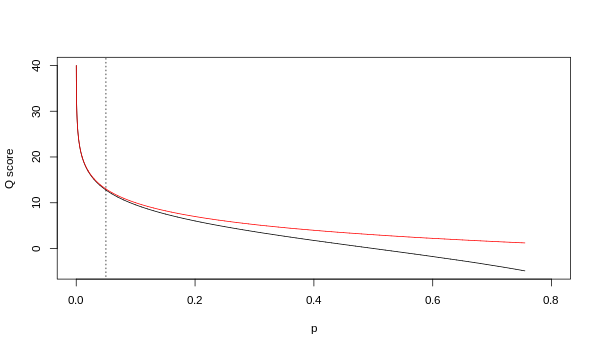
使用Sanger(红色)和Solexa(黑色)方程(如上所述)的Q和p之间的关系。 垂直虚线表示p = 0.05,或等同地Q≈13。
llumina流程1.4版用户指南(附录B,第122页)指出:“评分定义为:Q=10*log10(p/(1-p)),其中p是对应碱基判定的概率”。Illumina流程1.5版用户指南(第5页新增内容)对此进行了修改,“流程1.3版质量评分方案已更改为Phred评分方案,质量得分按Phred+64被转换为ASCII字符表示。”
编码
- Sanger格式可以使用ASCII 33~126字符表示0~93的Phred质量分数(尽管在原始read数据中,Phred质量分数很少超过60,但在组装或比对中可能有更高的分数)。这种方法也用于SAM格式中。根据seqanswers.com 上的公告,到2011年2月底,Illumina最新版本(1.8)的CASAVA流程将直接产生Sanger格式的fastq文件。
- Solexa/Illumina 1.0格式可以使用ASCII 59~126字符表示-5~62的Solexa/Illumina评分(原始read数据Solexa评分仅为-5~40)。
- 从Illumina 1.3版本开始,到Illumina 1.8版本,使用ASCII 64~126表示0~62的Phred质量分数(原始数据中Phred分数仅为0~40)。
- 从Illumina1.5版本开始,到Ilumina 1.8版本,Phred分数为0~2的含义略有不同。值0和1不在使用,值2(ASCII字符“B”)作为read片段结束的质量控制指示器。Illumina手册(第30页)内容:如果read以质量最差的一段(Q15或更低)结束,则片段中的所有质量值将被值2替换(ASCII字符“B”)…此Q2指标不能表示特定的错误率,而是表示read的特定最终部分不应用于进一步分析。此外,在Illumina 1.6版本之前,质量分数编码为“B”的碱基也可能在read内部发生,如下所示:
@HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1 TTAATTGGTAAATAAATCTCCTAATAGCTTAGATNTTACCTTNNNNNNNNNNTAGTTTCTTGAGATTTGTTGGGGGAGACATTTTTGTGATTGCCTTGAT +HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1 efcfffffcfeefffcffffffddf`feed]`]_Ba_^__[YBBBBBBBBBBRTT\]][]dddd`ddd^dddadd^BBBBBBBBBBBBBBBBBBBBBBBB
- 对于原始的read,质量分数的范围取决于所使用的技术和碱基判定标准,但对于最新的Illumina流程,通常会达到41.由于最大观察质量分数以前仅为40,因此各种脚本工具在遇到质量值大于40的数据时会中断。而对于经过过处理的read,质量分数可能会更高。例如:Illumina的Long Read Sequencing Service(以前的Moleculo)中read的质量分数为45。
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS..................................................... ..........................XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX...................... ...............................IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII...................... .................................JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ..................... LLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLL.................................................... !"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~ | | | | | | 33 59 64 73 104 126 0........................26...31.......40 -5....0........9.............................40 0........9.............................40 3.....9..............................41 0.2......................26...31........41 S - Sanger Phred+33, raw reads typically (0, 40) X - Solexa Solexa+64, raw reads typically (-5, 40) I - Illumina 1.3+ Phred+64, raw reads typically (0, 40) J - Illumina 1.5+ Phred+64, raw reads typically (3, 41) with 0=unused, 1=unused, 2=Read Segment Quality Control Indicator (bold) (Note: See discussion above). L - Illumina 1.8+ Phred+33, raw reads typically (0, 41)
色域
SOLiD数据,除了第一个位置,序列信息存储在色域中。质量值是Sanger格式的质量值,比对工具在质量值的首选版本上有所不同:其中一些包括主要和核苷酸的质量评分,而另一些则没有。序列read存档文件包含此质量分数。
模拟
压缩
质量分数占FASTQ格式所需磁盘空间(压缩前)的一般左右,因此压缩质量值可显著降低存储需求并加快测序数据的分析和传输。文献中提到的压缩方式主要无损和有损压缩。例如,有损压缩QualComp,无损压缩SCALCE、fastqz。
从Hiseq 2500开始,Illumina提供了将粗粒度质量输出到质量表的选项。分积分数直接从经验质量分数表中计算得出,该分数表与测序实验中所使用的硬件、软件和化学物质有关。
文件拓展名
FASTQ文件并没有标准的文件拓展名,但通常都是.fq或.fastq。
格式转换器
- Biopython version 1.51 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- EMBOSS version 6.1.0 patch 1 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- BioPerl version 1.6.1 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- BioRuby version 1.4.0 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- BioJava version 1.7.1 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- MAQ can convert from Solexa to Sanger (use this patch to support Illumina 1.3+ files).
- fastx_toolkit The included fastq_quality_converter program can convert Illumina to Sanger
命令行转换
FASTQ to FASTA format:
zcat input_file.fastq.gz | awk 'NR%4==1{printf ">%s\n", substr($0,2)}NR%4==2{print}' > output_file.fa
Illumina FASTQ 1.8 to 1.3
sed -e '4~4y/!"#$%&'\''()*+,-.\/0123456789:;<=>?@ABCDEFGHIJ/@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\\]^_`abcdefghi/' myfile.fastq # add -i to save the result to the same input file
Illumina FASTQ 1.3 to 1.8
sed -e '4~4y/@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\\]^_`abcdefghi/!"#$%&'\''()*+,-.\/0123456789:;<=>?@ABCDEFGHIJ/' myfile.fastq # add -i to save the result to the same input file
Illumina FASTQ 1.8 raw quality to binned quality (HiSeq Qtable 2.10.1, HiSeq 4000 )
sed -e '4~4y/!"#$%&'\''()*+,-.\/0123456789:;<=>?@ABCDEFGHIJKL/))))))))))----------77777<<<<<AAAAAFFFFFJJJJ/' myfile.fastq # add -i to save the result to the same input file
Illumina FASTQ 1.8 raw quality to clinto format (a visual block representation)
sed -e 'n;n;n;y/!"#$%&'\''()*+,-.\/0123456789:;<=>?@ABCDEFGHIJKL/▁▁▁▁▁▁▁▁▂▂▂▂▂▃▃▃▃▃▄▄▄▄▄▅▅▅▅▅▆▆▆▆▆▇▇▇▇▇██████/' myfile.fastq # add -i to save the result to the same input file
参考资料

 浙公网安备 33010602011771号
浙公网安备 33010602011771号