电子结构:能带和态密度计算
前面vasp简介已经说到电子结构计算总体分三步(有的也可以第23步一起计算),讲解了第一步,现在叙述第2、3步,用到的例子与前面提到的相同。
一、静态计算(Scf)
正常优化结构以后,进行静态计算重新建立文件夹,本人一般习惯将CONTCAR复制后重命名为POSCAR,将电荷密度和波函数以及其他4个基本输入文件复制到SCF文件夹当中,然后对INCAR进行一定的修改
- ISTART=1,代表现在从对读取已有的波函数和态密度进行参考计算,若保持为0则会从头开始计算;
NSW离子步为0,代表只运行一个离子步,此时IBRION=-1,如果不更改也会默认为-1;
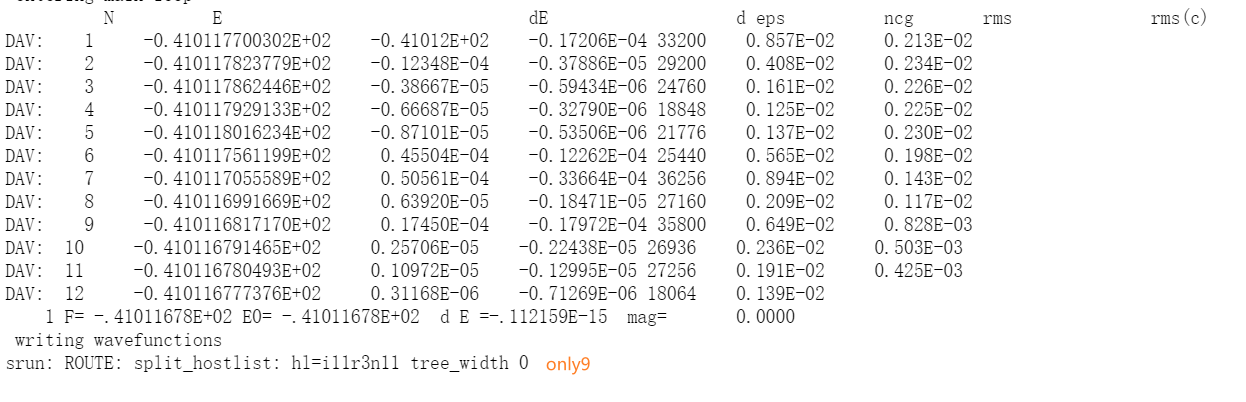
其他:关于离子步收敛的标准删除了,当然不删除在其他参数设置好了的情况下应该也不影响,同样的这里打开了波函数和电荷密度文件开关,这些都准备好了就可以进行静待计算了,计算完后下图:
二、能带结构计算
计算过程:静态计算完成以后对计算完后的四个基本输入文件和电荷密度、波函数文件以及提交任务脚本(前面未提到,需要根据自身设置)重新复制到新的文件夹中,命名为Band,在对INCAR进行修改,同时对能带计算的KPOINTS也要修改,这里本人使用VASPKIT自动生成,具体命令为"vaspkit>303",处理完成生成KPATH.in文件,将其重命名为KPOINTS,其内容见下图。INCAR以例子进行讲解: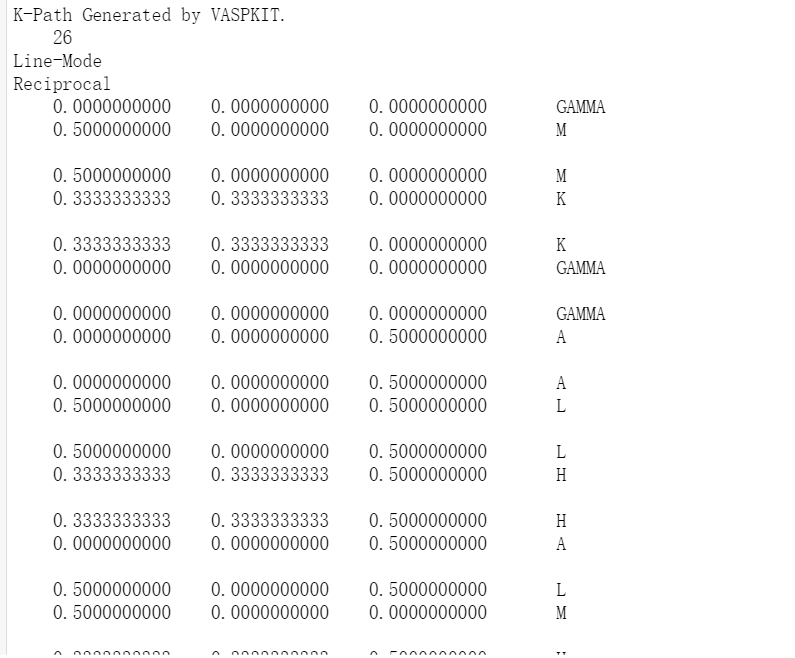
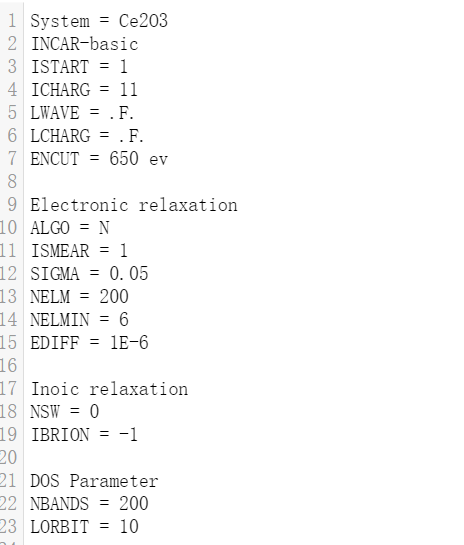
这里相较于静态计算增加了参数ICHARG=11,代表从已有的态密度和波函数文件读取信息;NBANDS和LORBIT,分别代表计算的能带数量和投影类型,NBANDS的选择也是根据先前计算的OUTCAR文件数据参考,使用'grep NBANDS OUTCAR'命令,取其1.5倍左右,可以尽可能取大一点;LORBIT=10代表只投影到元素轨道,取11可以投影到具体的原子轨道,观察不同能带由原子轨道的贡献情况。修改完成以后提交计算。
注意
计算能带时沿着特定的布里渊区进行积分,而不是整个布里渊区的均匀积分,因此参数ISMEAR=1或0,即使对于半导体也是如此;其次使用SIGMA=0.1左右提高精度
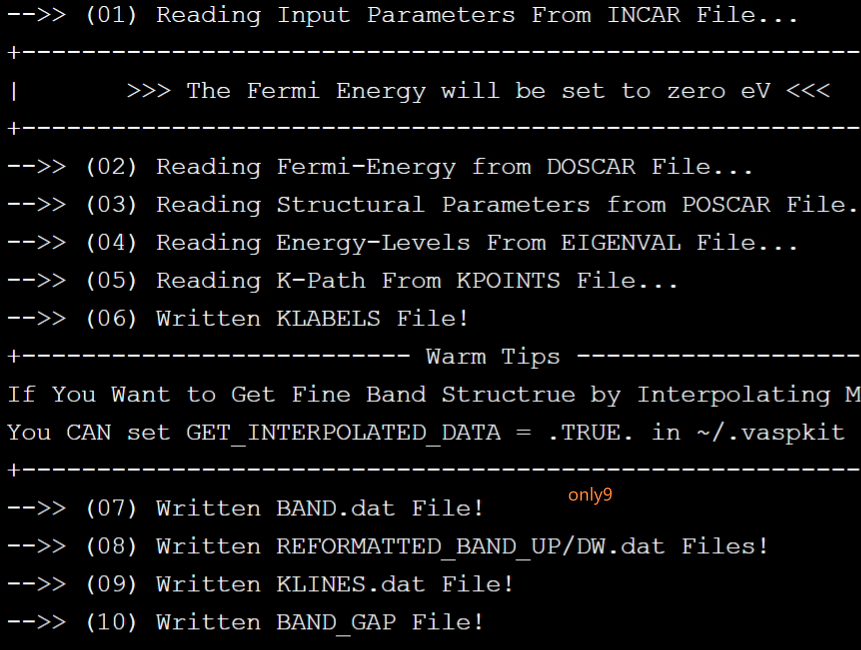
后处理:计算收敛以后,本人使用vaspkit相应的模块后处理,具体为"vaspkit |> 211",处理情况如下图,其中BAND.dat可以下载使用ORIGIN作图,BAND_GAPC查看带隙,KLABELS则是使用的的高对称点及其坐标。
使用Oringin作图如下图所示: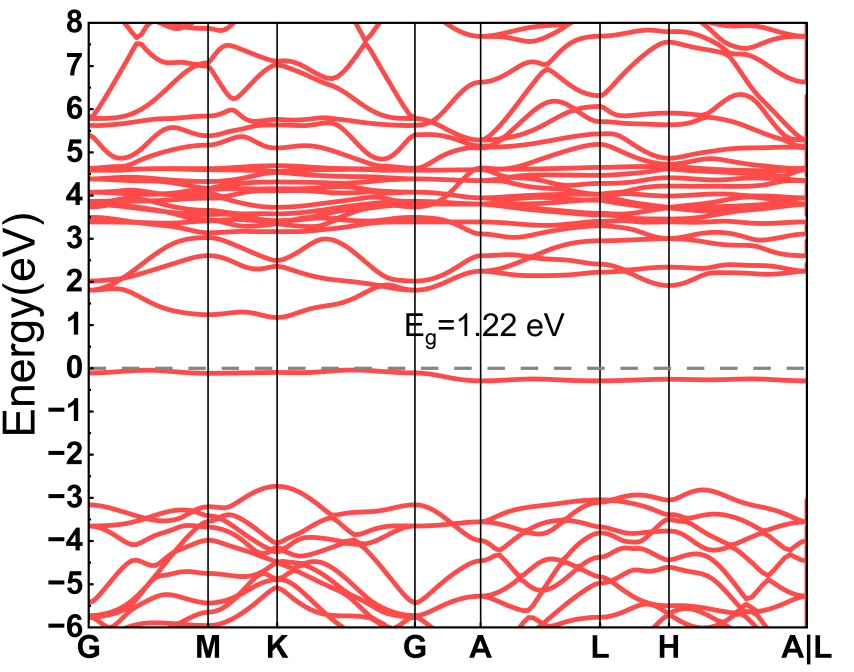
三、态密度计算
输入文件:主要的操作跟能带计算相似,输入文件当中KPOINTS保持不变,只是需要添加的参数略有不同,INCAR添加参数如下图:
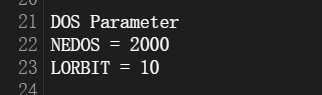
DEDOS参数本人暂不确定具体参照基准,一般设置1000左右,具体肯定根据体系,一般越大DOS图越精细和平滑,足够大以后曲线变化不大;LORBIT为10同样只投影到原子轨道,11则投影到原子分轨道。修改完后提交计算。
后处理:计算完后依然通过vaspkit处理,具体为'vaspkit>111'和"vaspkit>113"分别输出总的TDOS.dat和分的PDOS.dat,如果LORBIT=11时候可以选择特定原子的特定轨道态密度进行输出。将处理后的数据用Origin作图为: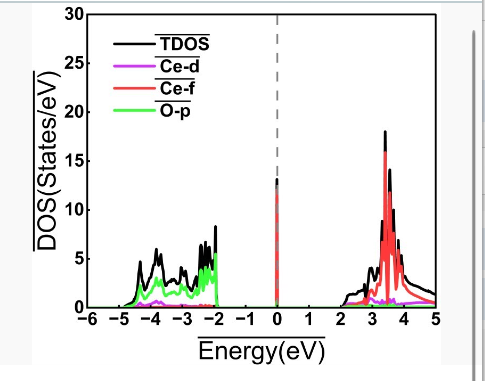
一般dos图分为自旋向上的自旋向下的部分,如果完全对称则总磁矩为0,所以本人只取了自旋向上的部分。如果两部分不对称则体系总磁矩不为0,这部分体系本人计算涉及较少。
tips:在每一次独立计算的vasp过程当中,如上述例子,能带和态密度其实时两个平行的计算分支,两者都是非自洽计算,但是对于vasp而言得分开建立两个文件夹分别计算,虽然计算能带时候也可以得出态密度相关文件,但官网说是不准确的,所以应该分开。当然对于某些平台也可以在第一步结构优化以后将静态计算和能带、态密度等等一同计算,所以具体视具体情况。。。。

 浙公网安备 33010602011771号
浙公网安备 33010602011771号